I’m very interested in the theme of using gene-gene graphs to guide and regularize learning, where it can be a remarkably effective technique to improve the efficacy of ML models on genes.
Much of the existing knowledge about gene function in various contexts can be described in terms of gene sets. They require some more intermediate modifications to guide and regularize learning. I’ll write about these here, to put them in one place for downstream applications.
As a prototype example, the GO is a controlled vocabulary for describing gene function – an indispensable tool for performing analyses over protein-coding genes. The GO is basically composed of gene sets corresponding to GO terms (nodes) and relationships between them (edges). This knowledge is essentially manually assembled, painstakingly from decades of experiments in the field. Building a gene-gene graph from the GO is a useful way to richly capture this knowledge.
I’ll cover how to build a gene-gene graph using GO. The first step is to get the relevant gene sets we need from GO.
Getting all GO gene sets
We’ll use the EnrichR API via the GSEAPy package to get gene sets corresponding to GO terms.
Here is a list of the protein-coding genes that we’ll be considering in this post.
CODE
import anndata, scanpy as scimport pandas as pd, matplotlib.pyplot as plt, timefrom sklearn import preprocessingsc.settings.verbosity =0%matplotlib inlineimport requests, os, numpy as npimport scipy, scipy.sparsegene_names_url ='https://raw.githubusercontent.com/kundajelab/coessentiality-browser/master/data/vizdf_GLS01_CO99.csv'gene_names = np.ravel(pd.read_csv(gene_names_url, header=0, index_col=None, sep="\t")['gene_names'])
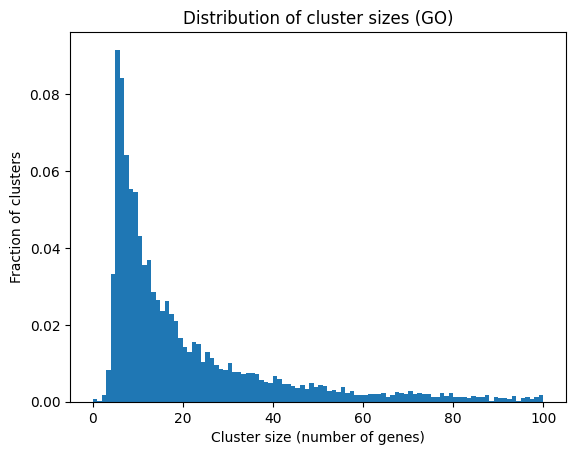
To get a basic idea of the gene sets in GO, we can plot the distribution of cluster sizes.
CODE
def membership_mat(genesets_dct, gene_names, file_path='memberships.npz'):""" Returns a sparse matrix of gene memberships to clusters. The matrix is of shape (n_genes, n_clusters), and is saved to disk as a sparse matrix. """ifnot os.path.exists(file_path):# Reverse the key-value mapping of the dictionary genesets_dict_rev = { g: [] for g in gene_names }for key, value in genesets_dct.items():for gene in value:if gene in genesets_dict_rev: genesets_dict_rev[gene].append(key) set_names =list(genesets_dct.keys()) nclusts =len(genesets_dct) clust_mmships = np.zeros((len(gene_names), nclusts))for i inrange(len(set_names)):if set_names[i] in genesets_dct: clust_mmships[np.isin(gene_names, genesets_dct[set_names[i]]), i] =1if i%250==0:print(i) clust_mmships = scipy.sparse.csr_matrix(clust_mmships) scipy.sparse.save_npz(file_path, clust_mmships)else: clust_mmships = scipy.sparse.load_npz(file_path)return clust_mmshipsclust_mmships_GO = membership_mat(go_dict, gene_names, file_path='GO_memberships.npz')plt.hist(np.sum(clust_mmships_GO.toarray(), axis=0), bins=100, density=True, cumulative=False, range=(0,100))plt.title("Distribution of cluster sizes (GO)")plt.xlabel("Cluster size (number of genes)")plt.ylabel("Fraction of clusters")plt.show()
We see that most of the gene sets are small, with a long tail of larger gene sets. So the modules being highlighted here are mostly relatively small and informative.
Data-driven gene sets
Data like gene expression can be used to partition genes into gene modules. One example of great functional and conceptual relevance is in the paper (Wainberg et al. 2021) (which I was involved in). The paper uses GO-derived gene sets as a crucial component to build a functionally relevant graph which faithfully recapitulates known biology, so it’s a great template to build upon. So I’ll delve deeper into some of the computational ideas from this paper.
The goal is to construct a network in which neighboring genes have similar functions. The most common way of constructing such a network is through co-expression analysis. But this has serious limitations with spurious correlations, because correlation is not causation.
A much better idea to construct functionally relevant gene networks is to use CRISPR knockout data instead of co-expression data. The idea here is that two genes may be functionally related if they are coessential – necessary for cell survival – in the same cell types. Therefore, performing a regression of coessentiality scores between a pair of genes gives a sensitive measure of functional similarity between the genes. And a gene-gene graph can be constructed from performing these regressions (through savvy implementation, they can be done in parallel on a laptop) through a generalized least squares (GLS) procedure.
I’ll write elsewhere about how to construct this graph, as it deserves a separate post. But for now, here’s code to load a precomputed version of the graph, roughly adapting associated code from (Wainberg et al. 2021).
CODE
def build_knn(mat, k=10, symmetrize_type='inclusive'):""" Build symmetric kNN graph, returning a sparse matrix. Parameters ---------- mat : numpy array Input matrix of similarities. k : int Number of nearest neighbors. symmetrize_type : str Type of symmetrization to use. * 'mutual': min(A, A^T), making degree \leq k * 'inclusive': max(A, A^T), making degree \geq k Returns ------- sparse_adj : scipy.sparse.csr_matrix Sparse adjacency matrix of the kNN graph. """ sparse_adj = scipy.sparse.csr_matrix(np.zeros(mat.shape))for i inrange(mat.shape[0]): matarr = mat[i,:] nbrs = np.argsort(matarr)[::-1][:k] # Highest k similarities sparse_adj[i, nbrs] =1.0#matarr[nbrs]if (symmetrize_type =='mutual'):return sparse_adj.minimum(sparse_adj.transpose())elif (symmetrize_type =='inclusive'):return sparse_adj.maximum(sparse_adj.transpose())else:print("Mode not yet implemented.")return sparse_adj
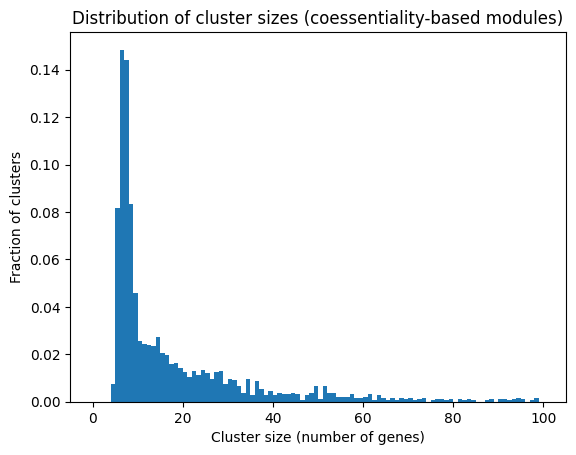
A clustering / community detection algorithm can be used to partition the graph into gene modules. This has already been done for the paper (Wainberg et al. 2021), and the resulting gene sets are loaded below.
CODE
# Read in clusterONE analysis results.def get_coess_module_dict(): clusterone_url ='https://raw.githubusercontent.com/kundajelab/coessentiality/master/clusterOne_clusters.tsv' cone_clusts = pd.read_csv(clusterone_url, sep="\t", index_col=0, header=0) clustdata = cone_clusts.iloc[:,10:].fillna(value='') genesets_dict = {}for i inrange(clustdata.shape[0]): a = clustdata.iloc[i,:].values genesets_dict[clustdata.index[i]] = a[a !='']return genesets_dictgenesets_dict = get_coess_module_dict()clust_mmships_coessentiality = membership_mat(genesets_dict, gene_names, file_path='clusterone_memberships.npz')plt.hist(np.sum(clust_mmships_coessentiality.toarray(), axis=0), bins=100, density=True, cumulative=False, range=(0,100))plt.title("Distribution of cluster sizes (coessentiality-based modules)")plt.xlabel("Cluster size (number of genes)")plt.ylabel("Fraction of clusters")plt.show()
Graphs from gene sets
The next step is to process these gene sets into a neighborhood-based matrix representing the gene sets.
Pairwise similarity between genes
One way to do this is by creating a graph gg_jaccsims where each node is a gene, and each edge is a Jaccard similarity measure between the GO terms of the two genes. The Jaccard similarity is the number of sets that both genes are in (“intersection”), divided by the number of sets that either gene is in (“union”).
CODE
def pairwise_jaccard_graph(input_memberships):""" Given a bunch of entities (like genes) and their binary memberships in some sets (like GO terms), construct graph of Jaccard similarities between the entities. Parameters ---------- input_memberships : scipy.sparse.csr_matrix Sparse matrix of set memberships. Each row is an entity, each column is a set. Returns ------- numpy.ndarray Matrix of pairwise Jaccard similarities. """ a = input_memberships.dot(input_memberships.T).toarray() tmp = np.add(np.add(-a.T, np.diagonal(a)).T, np.diagonal(a))return np.nan_to_num(np.divide(a, tmp))# Build and save pairwise Jaccard similarities between genes, according to the clusterings given.gg_jaccsims_GO = pairwise_jaccard_graph(clust_mmships_GO)gg_jaccsims_coessentiality = pairwise_jaccard_graph(clust_mmships_coessentiality)
/var/folders/5b/ps6ymxr90tj0jglr7hvc98zm0000gn/T/ipykernel_91723/703627734.py:17: RuntimeWarning: invalid value encountered in divide
return np.nan_to_num(np.divide(a, tmp))
There are stark differences between the data-derived gene sets and GO. The data-derived gene sets are significantly smaller and more specific, resulting in higher similarities between genes using our method above.
CODE
plt.hist(gg_jaccsims_GO[gg_jaccsims_GO >0], bins=150, cumulative=False, density=True, label='GO')plt.hist(gg_jaccsims_coessentiality[gg_jaccsims_coessentiality >0], bins=150, cumulative=False, density=True, label='Coessentiality')plt.title("Distribution of nonzero Jaccard similarities between genes")plt.xlabel("Jaccard similarity")plt.ylabel("Fraction of gene pairs")plt.legend()plt.show()
The graphs are not particularly sparse, so we sparsify this graph by building a symmetric kNN matrix from it.
/Users/akshay/opt/anaconda3/envs/env-chemML/lib/python3.10/site-packages/scipy/sparse/_index.py:146: SparseEfficiencyWarning: Changing the sparsity structure of a csr_matrix is expensive. lil_matrix is more efficient.
self._set_arrayXarray(i, j, x)
This is ultimately a graph which is much easier to deal with for computation, as I’ve written about elsewhere. By using the Jaccard similarity, we normalize for the fact that some genes are in many more GO terms than others.
Gene sets as neighborhoods
Here’s another way of building a graph from gene sets. Each gene set can be represented in the overall gene-gene graph by a clique – a subgraph where each gene in the gene set is connected to every other gene in it. Adding the resulting cliques together builds a graph that seamlessly deals with overlapping gene sets. When the gene sets are relatively small, as above, this resulting graph is also relatively sparse.
We normalize each graph by the number of nodes in the gene set, which also normalizes the added degrees of each node in the clique. We save the resulting graph for further use.
CODE
def create_clique_adj_matrix(master_list, active_list, weight='reciprocal'):""" Create clique composed of the elements in active_list that are in master_list. """ n =len(master_list) data, row_indices, col_indices = [], [], []# Create a dictionary for faster look-up active_list = np.intersect1d(master_list, active_list) gene_to_index = {gene: index for index, gene inenumerate(master_list)}for gene1 in active_list:for gene2 in active_list:if gene1 != gene2: row = gene_to_index[gene1] col = gene_to_index[gene2] row_indices.append(row) col_indices.append(col)if weight=='reciprocal': data.append(1.0/len(active_list))elif weight=='binary': data.append(1.0)return scipy.sparse.csr_matrix((data, (row_indices, col_indices)), shape=(n, n))def genesets_clique_matrix(geneset_dict, gene_names, file_path='clique_graph.npz'):""" Return dictionary mapping GO terms to their clique adjacency matrices. """ifnot os.path.exists(file_path): matrix_return = scipy.sparse.csr_matrix((len(gene_names), len(gene_names))) i =0for termname in geneset_dict: i +=1 matrix_return += create_clique_adj_matrix(gene_names, geneset_dict[termname]) scipy.sparse.save_npz(file_path, matrix_return)else: matrix_return = scipy.sparse.load_npz(file_path)return matrix_return
We can now process the graph to make it more useful for downstream analysis, by applying several data science techniques to it, following (Wainberg et al. 2021).
Mixing in a connected component
One potential issue with using the GO graph for learning is that it is not fully connected - there are many genes that are not connected to any other genes, because they don’t appear in GO’s gene modules.
CODE
all_genes_go_dict =list(set([item for sublist in [go_dict[k] for k in go_dict.keys()] for item in sublist]))num_genes_connected =len(np.intersect1d(gene_names, all_genes_go_dict))print("{} genes are in at least one GO gene set. {} are in no GO gene sets.".format( num_genes_connected, len(gene_names) - num_genes_connected))
14139 genes are in at least one GO gene set. 3495 are in no GO gene sets.
The same is true of the coessentiality-derived gene modules.
CODE
all_genes_go_dict =list(set([item for sublist in [genesets_dict[k] for k in genesets_dict.keys()] for item in sublist]))num_genes_connected =len(np.intersect1d(gene_names, all_genes_go_dict))print("{} genes are in at least one coessentiality-derived gene set. {} are in no coessentiality-derived GO gene sets.".format( num_genes_connected, len(gene_names) - num_genes_connected))
15374 genes are in at least one coessentiality-derived gene set. 2260 are in no coessentiality-derived GO gene sets.
One way to solve this issue is to use the fully connected graph derived from coessentiality data, and mix it in with the GO graph.
CODE
def coessentiality_GLS_graphs( file_path='GLS_pvals_10NN.npz', fname_GLS_p ='GLS_p.npy', GLS_p_url='https://mitra.stanford.edu/bassik/coessentiality/GLS_p.npy', min_logp=1000):""" Return sparse matrix of coessentiality (-log p) values between genes, calculated using GLS. """ifnot os.path.exists(fname_GLS_p): response = requests.get(GLS_p_url)withopen(fname_GLS_p, 'wb') as fp: fp.write(response.content) GLS_p = np.load(fname_GLS_p) res =-np.log(GLS_p) res[np.isinf(res)] = min_logpifnot os.path.exists(file_path): GLS_pvals_100 = build_knn(res, k=100, symmetrize_type='inclusive') GLS_pvals_10 = build_knn(GLS_pvals_100.toarray(), k=10, symmetrize_type='inclusive') scipy.sparse.save_npz(file_path, GLS_pvals_10) GLS_pvals_10 = scipy.sparse.load_npz(file_path)return GLS_pvals_10, res
Putting it all together, mixing the two graphs’ adjacency matrices together with some weighting, gives a function for getting a gene-gene graph.
This involves using diffusion maps to emphasize the more global coarse-scale structure of the graph, smoothing out high-frequency components to a degree such that the top few components capture most of the variance in the graph.
This is a useful technique for downstream learning, in the same way that PCA-based dimensionality reduction is used in computational workflows. In addition, it can help to provide a more visualizable representation of the input graph.
CODE
def compute_transitions(adj_mat, alpha=1.0, sym=True):"""Compute symmetrized transition matrix. alpha : The exponent of the diffusion kernel. * 1 = Laplace-Beltrami (density)-normalized [default]. * 0.5 = normalized graph Laplacian (Fokker-Planck dynamics, cf. "Diffusion maps, spectral clustering and eigenfunctions of Fokker-Planck operators" NIPS2006). * 0 = classical graph Laplacian. """ similarity_mat = symmetric_part(adj_mat) dens = np.asarray(similarity_mat.sum(axis=0)) # dens[i] is an estimate for the sampling density at point i. K = scipy.sparse.spdiags(np.power(dens, -alpha), 0, similarity_mat.shape[0], similarity_mat.shape[0]) W = scipy.sparse.csr_matrix(K.dot(similarity_mat).dot(K)) z = np.sqrt(np.asarray(W.sum(axis=0)).astype(np.float64)) # sqrt(density) Zmat = scipy.sparse.spdiags(1.0/z, 0, W.shape[0], W.shape[0])return Zmat.dot(W).dot(Zmat) if sym else Zmat.power(2).dot(W)def compute_eigen(adj_mat, n_comps=2, sym=True):""" Compute eigendecomposition of sparse transition kernel matrix. (Computing in 64-bit can matter for analysis use beyond just visualization!). sym indicates that the transition matrix should be symmetrized.NOTE: use np.linalg.eigh if we want to support non-sparse matrices. """if sym: eigvals, evecs = scipy.sparse.linalg.eigsh(adj_mat.astype(np.float64), k=n_comps)else:# eigvals, evecs = scipy.sparse.linalg.eigs(adj_mat.astype(np.float64), k=n_comps) # DON'T USE without further thresholding: complex-number underflow issues evecs, eigvals, _ = scipy.sparse.linalg.svds(adj_mat.astype(np.float64), k=n_comps)#eigvals, evecs = eigvals.astype(np.float32), evecs.astype(np.float32) sorted_ndces = np.argsort(np.abs(eigvals))[::-1]return eigvals[sorted_ndces], evecs[:, sorted_ndces]""" Returns the symmetrized version of the input matrix. (The anti-symmetrized version is A - symmetric_part(A) .) """def symmetric_part(A):return0.5*(A + A.T)"""Compute diffusion map embedding.sym_compute: Whether to compute the SVD on a symmetric matrix.sym_return: Whether to return the SVD of the symmetrized transition matrix.Assumes that {sym_return => sym_compute} holds, i.e. doesn't allow sym_compute==False and sym_return==True.Returns (n_comps-1) components, excluding the first which is the same for all data."""def diffmap_proj( adj_mat, t=None, min_energy_frac=0.95, n_dims=None, # Number of dims to return n_comps=None, # Number of comps to use in computation. return_eigvals=False, embed_type='diffmap', sym_compute=True, sym_return=False):if n_comps isNone: # When data are high-d dimensional with log2(d) \leq 14-16 as for scRNA, 2K eigenvector computation is tolerably fast; change otherwise n_comps =min(2000, adj_mat.shape[0]-1)if n_dims isNone: n_dims = n_comps -1 eigvals, evecs = compute_eigen(compute_transitions(adj_mat, sym=sym_compute), n_comps=n_comps, sym=sym_compute)if sym_compute: evecs_sym = evecs evecs_unsym = np.multiply(evecs, np.outer(1.0/evecs[:,0].astype(np.float64), np.ones(evecs.shape[1])))else: evecs_unsym = evecsif sym_return:ifnot sym_compute:returnNone eigvecs_normalized = preprocessing.normalize(np.real(evecs_sym), axis=0, norm='l2')else: eigvecs_normalized = preprocessing.normalize(np.real(evecs_unsym), axis=0, norm='l2')if t isNone: # Use min_energy_frac to determine the fraction of noise variance t = min_t_for_energy(eigvals, n_dims+1, min_energy_frac) frac_energy_explained = np.cumsum(np.power(np.abs(eigvals), t)/np.sum(np.power(np.abs(eigvals), t)))[n_dims]print("{} dimensions contain about {:.2f} fraction of the variance in the first {} dimensions (Diffusion time = {:.3f})".format( n_dims+1, frac_energy_explained, n_comps, t))if embed_type=='naive': all_comps = eigvecs_normalizedelif embed_type=='diffmap': all_comps = np.power(np.abs(eigvals), t) * eigvecs_normalizedelif embed_type=='commute': # Check the correctness! all_comps = np.power((1-np.abs(eigvals)), -t/2) * eigvecs_normalizedifnot return_eigvals:return all_comps[:,1:(n_dims+1)] # Return n_dims dimensions, skipping the first trivial one.else:return (all_comps[:,1:(n_dims+1)], eigvals) # Return the eigenvalues as well.def min_t_for_energy(eigvals, desired_dim, min_energy_frac, max_t=None):# Calc upper bound for t principal eigengap (if g=lbda1/lbda2, then g^t < 100 implies t < log(100)/log(g) ). Don't change unless you know what you're doing!if max_t isNone: max_t = np.log(100)/np.log(max(eigvals[0]/eigvals[1], 1.01)) f =lambda t: (np.sum(heat_eigval_dist(eigvals, t)[:desired_dim]) - min_energy_frac)if f(0)*f(max_t) >=0: # since f is always nondecreasing this means the zero isn't in the intervalreturn max_t if f(0) <0else0return scipy.optimize.brentq(f, 0, max_t)def heat_eigval_dist(eigvals, t):return np.power(np.abs(eigvals), t)/np.sum(np.power(np.abs(eigvals), t))
41 dimensions contain about 0.90 fraction of the variance in the first 100 dimensions (Diffusion time = 95.917)
Diffusion components computed. Time: 4.27
50 dimensions contain about 1.00 fraction of the variance in the first 50 dimensions (Diffusion time = 0.000)
Laplacian eigenmap computed. Time: 1.78 sec.
Verify that the overall graph includes known gene modules
Read modules highlighted in the paper.
CODE
url_modules ='https://www.ncbi.nlm.nih.gov/pmc/articles/PMC8763319/bin/NIHMS1768167-supplement-Supp_Data_2.zip'import gzip, requestsfrom zipfile import ZipFilefile_path ='NIHMS1768167-supplement-Supp_Data_2.zip'response = requests.get(url_modules)withopen(file_path, 'wb') as fp: fp.write(response.content)# opening the zip file in READ mode with ZipFile(file_path, 'r') aszip: # printing all the contents of the zip file zip.printdir() # extracting all the files print('Extracting all the files now...') zip.extractall() print('Done!') xl = pd.ExcelFile('Supplementary_Data_2.xlsx')print(xl.sheet_names)ref_modules = xl.parse('Manual Annotation References')module_dict = {}module_delims =list(np.where(~pd.isna(ref_modules['Module name']))[0]) + [ref_modules.shape[0]]for i inrange(len(module_delims)-1): these_genes_df = ref_modules.iloc[module_delims[i]:module_delims[i+1], :] module_dict[these_genes_df.iloc[0, :2]['Module name']] = np.ravel(these_genes_df['Gene name'])
File Name Modified Size
Supplementary_Data_2.xlsx 2021-03-05 15:28:02 11721905
Extracting all the files now...
Done!
['Co-essential Modules', 'Manual Annotation References']
CODE
for m in module_dict: ann_heat.obs['selected'] = np.isin(gene_names, module_dict[m]).astype(int) arrsize = ann_heat.obs['selected']*150+4#sc.pl.umap(ann_heat, color='selected', title=m, size=arrsize, cmap='viridis')
First, we need some utility code to deal with gene sets. This computes similarities between genes based on their membership in gene sets. Given a particular universe of gene sets, the similarity between two genes is the Jaccard index between their set memberships.
CODE
ann_heat.obs.index = ann_heat.obs['gene_names']mods = ann_heat.obs['gene_names'].copy()for m in module_dict: mods[module_dict[m]] = mann_heat.obs['module'] = modsfor m in module_dict:print(m, (ann_heat[module_dict[m]].obsp['connectivities'] >0).mean())
ann_heat.obs.index = ann_heat.obs['gene_names']mods = ann_heat.obs['gene_names'].copy()for m in module_dict: mods[module_dict[m]] = mann_heat.obs['module'] = modsfor m in module_dict:print(m, (ann_heat[module_dict[m]].obsp['connectivities'] >0).mean())
ann_heat.obs.index = ann_heat.obs['gene_names']ann_heat.obs['module'] = ann_heat.obs['gene_names']for m in module_dict: ann_heat[module_dict[m]].obs['module'] = mfor m in module_dict:print(m, (ann_heat[module_dict[m]].obsp['connectivities'] >0).mean())
GO information is tightly related to other indirect ways of getting at gene function. The interplay between gene regulation and drug responses is a rich source of information about gene function. These sources are particularly prevalent in medicine and can be very useful in drug development.
The paper (Chandak, Huang, and Zitnik 2023) builds a knowledge graph with multiple types of entities for this purpose. This provides a useful way to contextually view the relationships between genes, drugs, and diseases.
CODE
import pandas as pdprimekg_df = pd.read_csv('primeKG/kg.csv')primekg_df[primekg_df['display_relation'] =='interacts with']['y_type'].value_counts()primekg_df#['x_type'].value_counts()
relation
display_relation
x_index
x_id
x_type
x_name
x_source
y_index
y_id
y_type
y_name
y_source
0
protein_protein
ppi
0
9796
gene/protein
PHYHIP
NCBI
8889
56992
gene/protein
KIF15
NCBI
1
protein_protein
ppi
1
7918
gene/protein
GPANK1
NCBI
2798
9240
gene/protein
PNMA1
NCBI
2
protein_protein
ppi
2
8233
gene/protein
ZRSR2
NCBI
5646
23548
gene/protein
TTC33
NCBI
3
protein_protein
ppi
3
4899
gene/protein
NRF1
NCBI
11592
11253
gene/protein
MAN1B1
NCBI
4
protein_protein
ppi
4
5297
gene/protein
PI4KA
NCBI
2122
8601
gene/protein
RGS20
NCBI
...
...
...
...
...
...
...
...
...
...
...
...
...
8100493
anatomy_protein_absent
expression absent
66747
4720
anatomy
cerebellar vermis
UBERON
5259
140
gene/protein
ADORA3
NCBI
8100494
anatomy_protein_absent
expression absent
63824
1377
anatomy
quadriceps femoris
UBERON
58254
105378952
gene/protein
KLF18
NCBI
8100495
anatomy_protein_absent
expression absent
63826
1379
anatomy
vastus lateralis
UBERON
58254
105378952
gene/protein
KLF18
NCBI
8100496
anatomy_protein_absent
expression absent
64523
2084
anatomy
heart left ventricle
UBERON
58254
105378952
gene/protein
KLF18
NCBI
8100497
anatomy_protein_absent
expression absent
67302
5384
anatomy
nasal cavity epithelium
UBERON
58254
105378952
gene/protein
KLF18
NCBI
8100498 rows × 12 columns
References
Chandak, Payal, Kexin Huang, and Marinka Zitnik. 2023. “Building a Knowledge Graph to Enable Precision Medicine.”Scientific Data 10 (1): 67.
Wainberg, Michael, Roarke A Kamber, Akshay Balsubramani, Robin M Meyers, Nasa Sinnott-Armstrong, Daniel Hornburg, Lihua Jiang, et al. 2021. “A Genome-Wide Atlas of Co-Essential Modules Assigns Function to Uncharacterized Genes.”Nature Genetics 53 (5): 638–49.