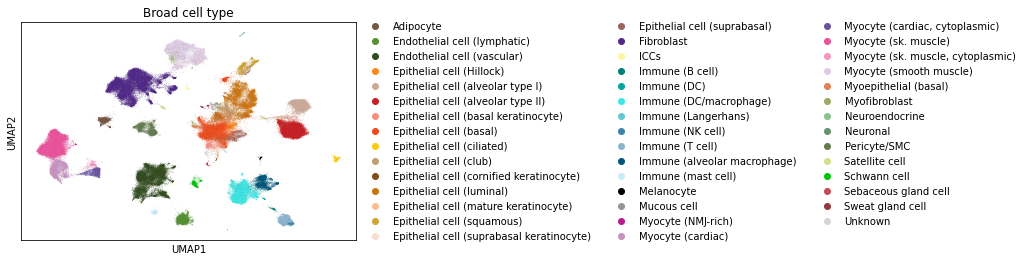As we’ve written elsewhere as above, building a data-driven interactive browser can unlock a lot of potential in displaying contextually toggled layers of information to users. Building this browser involves working with variables that are defined by the user interactively. The potential of this workflow can be realized through interactive computation that is driven by user selections.
This series of posts is developing and expanding a general, performant interactive data browser to explore the theme of interactive computation.
This particular post lays the groundwork for us to try some powerful interactive algorithms, by focusing on helping the user to select a subset of observations interactively, using the existing clustering and visualization capabilities built in the previous posts in this series.
Loading the previous browser
We first load the previously created interactive browser, as described in the previous post in the series. That post covers how to build a basic interactive interface with interactive clustering capabilities deployed in a heatmap. The code to run the browser is imported from the relevant file produced in that previous post.
CODE
ibrowser_path ="../../files/dataviz/interactive_browser_2.py"from importlib.machinery import SourceFileLoaderVE_browser = SourceFileLoader("VE_browser", ibrowser_path).load_module()# now we can importfrom VE_browser import*
---------------------------------------------------------------------------FileNotFoundError Traceback (most recent call last)
Cell In[1], line 4 1 ibrowser_path ="../../files/dataviz/interactive_browser_2.py" 3fromimportlib.machineryimport SourceFileLoader
----> 4 VE_browser =SourceFileLoader("VE_browser",ibrowser_path).load_module() 6# now we can import 7fromVE_browserimport*
File <frozen importlib._bootstrap_external>:548, in _check_name_wrapper(self, name, *args, **kwargs)
File <frozen importlib._bootstrap_external>:1063, in load_module(self, fullname)
File <frozen importlib._bootstrap_external>:888, in load_module(self, fullname)
File <frozen importlib._bootstrap>:290, in _load_module_shim(self, fullname)
File <frozen importlib._bootstrap>:719, in _load(spec)
File <frozen importlib._bootstrap>:688, in _load_unlocked(spec)
File <frozen importlib._bootstrap_external>:879, in exec_module(self, module)
File <frozen importlib._bootstrap_external>:1016, in get_code(self, fullname)
File <frozen importlib._bootstrap_external>:1073, in get_data(self, path)FileNotFoundError: [Errno 2] No such file or directory: '../../files/dataviz/interactive_browser_2.py'
Like a previous post, we are rendering a new type of data, so the workflow will be structured in two parts:
Preprocessing script: Given a dataset of chemicals, writes a dataframe that’s ready to be viewed by a front-end.
Interactive application: Given a dataframe, view it interactively in a browser.
Single-cell gene expression data
In addition to the applications in visualizing chemical space, such browsers have started to enter the toolbox for various biological and chemical datasets, particularly as the use of learned embeddings has exploded in recent years. A fruitful field of applications of such browsers, and the interactive algorithms they enable, is single-cell data.
Here, we demonstrate on single-cell data from the GTEx consortium, a large worldwide effort profiling gene expression across tissues.
This is a beautiful dataset to demonstrate systems biology on single-cell data, as in the recent paper introducing it (Eraslan et al. 2022). It is also very good to demonstrate new algorithms, as it is large (>200K cells and >17K genes) and comes well-annotated and preprocessed. The data are processed by a documented pipeline, which learns representations that attempt to correct for nuisance variation and batch effects.
This dataset is particularly notable for being the largest dataset of its kind using a single-nuclear RNA-seq (snRNA-seq) protocol, which attempts to isolate each cell nucleus, and sample unbiasedly from the RNA transcripts within it. This contrasts with more common and cheaper single-cell (scRNA-seq) protocols, which try to sample from the nucleus and cytoplasm (interior material) of each cell. This makes it much trickier to isolate the cells in scRNA-seq than snRNA-seq. The snRNA-seq dataset we will use here takes advantage of these characteristics, using cryogenically frozen tissues collected earlier by the same GTEx consortium, as detailed here.
As we can see, there are many cell types, whose annotations broadly match the cluster structure revealed by nonparametric dimensionality reduction and projection tools - in this case, UMAP (Becht et al. 2019).
There are ~209k observations and ~17k features, which is impossible to visualize all at once. So we need to do a little preprocessing to display a subset of this dataset interactively.
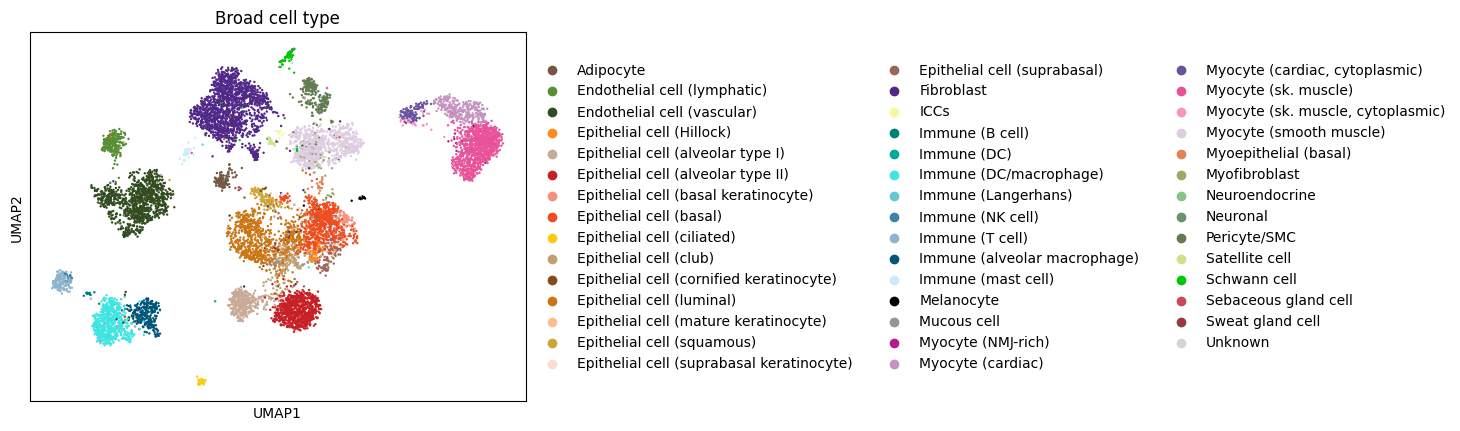
The approach I cover here is to subsample down to 10k cells, which is typically a nice number for the browser for visualization- and perception-based reasons.
Selecting a subset of smoothed data
The standard way to subset rows is to randomly subsample observations. This is easy to implement, but can lead to unacceptable levels of sparsity in the data. At the extremes, this is obvious; for instance, sampling just a hundred observations from our 200k risks missing entire observation clusters and the features uniquely found in them.
In an attempt to make up for this, the needs of the situation often dictate a more sophisticated method of subsampling, sometimes accompanied by a nonparametric smoothing of the data so as not to lose signal with sparse data. We precede things by a nonparametric smoothing step that mixes in a little of each observation’s neighbors for each feature. Since an observation’s neighbors typically have similar feature representations, this respects the initial representations of the data, while making it much less sparse. And it’s just one matrix multiplication, typically of two sparse matrices.
So the workflow I demonstrate here is:
(Optional) Smooth data using neighborhood structure
Subsample 10k observations and recompute neighborhood graph, using the learned representation that Eraslan et al. used in their analysis (Eraslan et al. 2022)
python(53546) MallocStackLogging: can't turn off malloc stack logging because it was not enabled.
/opt/anaconda3/envs/env-dash/lib/python3.10/site-packages/tqdm/auto.py:21: TqdmWarning: IProgress not found. Please update jupyter and ipywidgets. See https://ipywidgets.readthedocs.io/en/stable/user_install.html
from .autonotebook import tqdm as notebook_tqdm
Neighbors computed. Time: 23.66276979446411
The overall cluster structure (coarse close-range topology) of the subsample is the same, which is one indication1 that we can proceed with the subsample, and our newly computed neighborhood graph.
Writing the dataset to disk
The dataset can be written to disk for future analyses, after adding metadata columns representing its UMAP coordinates.
The modifications made to the code in this post ultimately result in an improved browser, the code for which is available here. Setting it up an environment will deploy the app.
Browser in action
As you can see, we can do several layers of hierarchical clustering, zooming into a few cells out of 10k, with just a few clicks and hardly any executive function!
Additional changes
In changing datasets out of chemical space, we also make a couple of other modifications.
Removing chemical functionality: The chemical images no longer need to be displayed, which entails removing the html <img> component chem2D-image and its dependencies; they are pretty modular and can be added back if needed.
Handling anndata objects: Some changes were made to update_heatmap() and to display_heatmap_cb(), as the object to be rendered is now an anndata object instead of a pandas dataframe. One particular change of note is to interesting_feat_ndces(), to handle sparse data. The variances are now calculated in a particular way that does not involve constructing a dense matrix as an intermediate step.
Color schemes and normalization: Strange things can happen when the wrong colormap is used: a discrete colormap for continuous ordinal data, for example. This is important enough to discuss separately in detail. We’ve already seen a lot of the work done, in some of the colormap code posted in the first post.
Selecting subclusters of the data from the heatmap
The heatmap, with its automatic coclustering, can be used as a vitally important tool in scaling things up. It depicts the structure of the data in a more informative way than a simple 2D projection like PCA or UMAP.
A really common interactive task to select a group of observations on the heatmap, and iteratively refine this subselection.
A key tool to do that is to allow a way to easily select a subset of rows from the heatmap. I do this by anchoring a selectable point alongside each row in the heatmap, constituting a parallel scatterplot whose state is kept linked to the main one in the browser.
The code for this is fairly boilerplate, implementing the logic of subset selection. I’ve included it below. A few notes for the interested:
A couple of comments bear mention.
The arguments include the dataframe in question (data_df), and the column in it that’s being used as the scatterplot color variable (color_var), both of which are used in the original scatterplot. The other required argument (hm_point_names) contains the names of the
If there are 30 or fewer points being displayed, text labels will be shown alongside the points. This is a somewhat arbitrary choice that makes things easier when users do narrow down to a small subcluster of data.
CODE
def hm_row_scatter(anndata_obj, color_var, hm_point_names, num_of_category=0, bg_marker_size=params['bg_marker_size_factor']): df_data = anndata_obj.obs num_colors_needed =len(np.unique(df_data[color_var])) colorscale_list = get_discrete_cmap(num_colors_needed) row_scat_traces = [] hmscat_mode ='markers'# Decide if few enough points are around to display row labelsiflen(hm_point_names) <=30: hmscat_mode ='markers+text' cnt =0for idx in np.unique(df_data[color_var]): val = df_data.loc[df_data[color_var] == idx, :] point_ids_this_trace =list(val.index) hm_point_where_this_trace = np.isin(hm_point_names, point_ids_this_trace) hm_point_names_this_trace = hm_point_names[hm_point_where_this_trace] num_in_trace =len(hm_point_names_this_trace) hm_point_ndces_this_trace = np.where(hm_point_where_this_trace)[0] # this trace's row indices in heatmap y_coords_this_trace = np.arange(len(hm_point_names))[hm_point_ndces_this_trace]# At this point, rows are sorted in order of co-clustering. trace_color = colorscale_list[cnt] cnt +=1 pt_text = ["{}".format(point_ids_this_trace[i]) for i inrange(len(point_ids_this_trace))] new_trace = {'name': str(idx), 'x': np.ones(num_in_trace)*-1*num_of_category, 'y': y_coords_this_trace, 'xaxis': 'x2', 'hoverinfo': 'text', 'text': [x for x in hm_point_names_this_trace], 'mode': hmscat_mode, 'textposition': 'middle left', 'textfont': hm_font_macro, 'marker': {'size': bg_marker_size, 'opacity': params['marker_opacity_factor'], 'symbol': 'circle', 'color': trace_color }, 'selected': style_selected, 'type': 'scatter' } row_scat_traces.append(new_trace)return row_scat_traces
To understand what this is doing, some key code was actually introduced in a previous post, though I didn’t comment on it much. The code in question is from the main heatmap creation code, in display_heatmap_cb, creating an alternate x-axis.
This creates the auxiliary scatterplot axis and physically links it to the main one.
Restructuring control flow
Finally, a few precise changes have to be made to the control flow to correctly link the new heatmap’s associated scatterplot to the rest of the interface.
The new heatmap’s scatterplot’s selected data is linked to the main scatterplot.
Similarly, landscape-plot.selectedData is added as an input dependency of landscape-plot.figure, so that the selected data update on the main scatterplot.
A displayed component num-selected-counter is added to count how many points are selected, and display the result to the user. This occupies little space, and can be difficult to gauge even within an order of magnitude otherwise.
After establishing a way to see the data in the first post, and a way to see the substructure of that data in heatmap form in the second post, we’ve done two things here:
Switch to a larger single-cell dataset and view its features.
Establish a way to select subsets using the interactively viewed substructure.
Next, I’ll share something I’ve been anticipating for a while - building some simple methods for interactive visualization into our browser.
This sets us up perfectly to do truly interactive computation in the browser, because supervised learning and other advanced algorithms run at interactive speeds on 10k observations. Future posts will head in this direction.
Bonus changes: refactoring coclustering for sparse data
On truly large, sparse data, we’d like the algorithm to be blazing fast. Such data result in very sparse graphs, which can be dealt with efficiently to find spectral cuts as per the algorithm. It’s just a matter of rewriting the coclustering algorithm code from scikit-learn and plugging the new implementation into our interface.
We are looking at refactoring just a small part of the scikit-learn code. But rather than accessing private parts of the code piecemeal, I’ll opt here to rewrite it explicitly - it’s not long, and quite insightful for future use.
def _fit(self, X): normalized_data, row_diag, col_diag = _scale_normalize(X) n_sv =1+int(np.ceil(np.log2(self.n_clusters))) u, v =self._svd(normalized_data, n_sv, n_discard=1) z = np.vstack((row_diag[:, np.newaxis] * u, col_diag[:, np.newaxis] * v)) _, labels =self._k_means(z, self.n_clusters) n_rows = X.shape[0]self.row_labels_ = labels[:n_rows]self.column_labels_ = labels[n_rows:]self.rows_ = np.vstack([self.row_labels_ == c for c inrange(self.n_clusters)])self.columns_ = np.vstack( [self.column_labels_ == c for c inrange(self.n_clusters)] )
This consists of a few steps, starting from the \((n \times d)\) data matrix \(X\):
The data matrix \(X\) is normalized: if the row sums of \(X\) are arranged in a \((n \times n)\) diagonal matrix \(R\) and the column sums are in a \((d \times d)\) diagonal matrix \(C\), the data are normalized according to \[X' := R^{-1/2} X C^{-1/2} \] This is the normalization function _scale_normalize()here.
The normalized data matrix’s top few left and right eigenvectors are computed and normalized.
The computed eigenvectors are clustered (by k-means) to get the co-clusters, and thereby ordered for display. This usage of eigenvector-based representations of the matrix’s respective rows and columns is akin to spectral clustering.
The old code’s implementation runs fast on sparse data, since it uses SVD routines which scale well when run on sparse data.
New code
We replace the above function by porting each step from sklearn:
The data matrix \(X\) is normalized: This is basically unchanged from sklearn’s way of doing it, _scale_normalize()here. The main difference is that we expect the input matrix to be nonnegative.
CODE
def scale_normalize(X):""" Normalize nonnegative ``X`` by scaling rows and columns independently. Returns the normalized matrix and the row and column scaling factors. """ row_diag = np.asarray(1.0/ np.sqrt(X.sum(axis=1))).squeeze() col_diag = np.asarray(1.0/ np.sqrt(X.sum(axis=0))).squeeze() row_diag[np.isinf(row_diag)] =0 col_diag[np.isinf(col_diag)] =0 row_diag = np.where(np.isnan(row_diag), 0, row_diag) col_diag = np.where(np.isnan(col_diag), 0, col_diag)if scipy.sparse.issparse(X): n_rows, n_cols = X.shape r = scipy.sparse.dia_matrix((row_diag, [0]), shape=(n_rows, n_rows)) c = scipy.sparse.dia_matrix((col_diag, [0]), shape=(n_cols, n_cols)) an = r * X * celse: an = row_diag[:, np.newaxis] * X * col_diagreturn an, row_diag, col_diag
The normalized data matrix’s top few left and right eigenvectors are computed and normalized. This involves using a carefully chosen SVD routine, sklearn.utils.extmath.randomized_svd(), which uses state-of-the-art SVD methods that do the computations in a nearly optimal low-rank subspace (Halko, Martinsson, and Tropp 2011). Other routines from scipy.sparse.linalg can be used as well for the same purpose, namely svds(), and eigsh() from the same library for symmetric matrices \(A\).
CODE
from sklearn.utils.extmath import randomized_svddef spectral_coembed( X, n_clusters=25, n_components=2, n_discard=1):""" Args: X: (n x d) data matrix used, n observations by d features. Returns: Spectral coembedding of (n + d) rows and columns of X; first the rows, then the columns. Uses (n_components - n_discard) components. """ normalized_data, row_diag, col_diag = scale_normalize(X) n_sv =1+int(np.ceil(np.log2(n_clusters))) if n_components isNoneelse n_components u, _, vt = randomized_svd(normalized_data, n_sv) u = u[:, n_discard:] v = vt[n_discard:].T z = np.vstack((row_diag[:, np.newaxis] * u, col_diag[:, np.newaxis] * v))return z
The computed rows/columns are ordered for display using the first coordinate of their representation, i.e. the eigenvector corresponding to the second-largest eigenvalue.
CODE
from sklearn.cluster import KMeansdef cocluster_from_embedding(X, n_clusters=25, cluster=True):""" Args: X: (n x d) data matrix used, n observations by d features. Returns: Indices to display the matrix with. """ z = spectral_coembed(X, n_clusters=n_clusters) n_rows = X.shape[0]if cluster: kmodel = KMeans(n_clusters) kmodel.fit(z) labels = kmodel.labels_ row_labels_ = labels[:n_rows] column_labels_ = labels[n_rows:]return np.argsort(row_labels_), np.argsort(column_labels_)else: fiedler_vec = z[:, 0]return np.argsort(fiedler_vec[:n_rows]), np.argsort(fiedler_vec[n_rows:])
This function cocluster_from_embedding is now callable by the main coclustering function the interface is using, as shown below. (I also included, and show here, a small tweak to select a default number of clusters based on the dimensions of the matrix.)
Finally, we do slightly change the heatmap rendering in the function display_heatmap_cb(), making a couple of accommodations for the changing configuration options.
Becht, Etienne, Leland McInnes, John Healy, Charles-Antoine Dutertre, Immanuel WH Kwok, Lai Guan Ng, Florent Ginhoux, and Evan W Newell. 2019. “Dimensionality Reduction for Visualizing Single-Cell Data Using UMAP.”Nature Biotechnology 37 (1): 38–44.
Halko, Nathan, Per-Gunnar Martinsson, and Joel A Tropp. 2011. “Finding Structure with Randomness: Probabilistic Algorithms for Constructing Approximate Matrix Decompositions.”SIAM Review 53 (2): 217–88.
Footnotes
In general, a 2D projection like UMAP is not a good way to reliably capture structure in a plot by eye. Straight lines, angles, and distances can be very deceptive (this is largely true even when reducing 3D to 2D, ‘viewing from an angle’). But it’s what we have, and pretty good for close-range cluster structure, which gets warped less by all the nonlinear shenanigans.↩︎