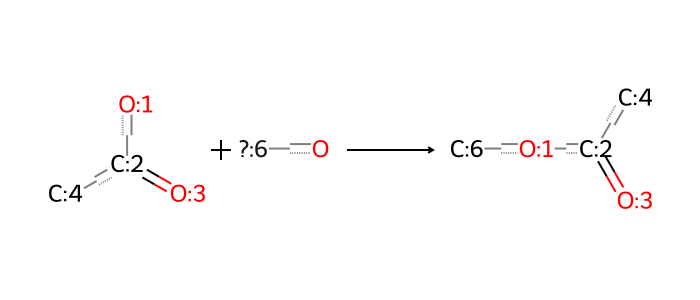This post builds on another post which demonstrates the basics of using SMARTS to easily put together fragments in silico. That post, which should be read first, shows a basic workflow for combinatorial synthesis – how to move from fragments to a fully synthesized structure. Those fragments can be procured from several sources like ready-to-synthesize catalogs. In this post, we explore a duality between decomposition and synthesis that can fully be exploited in silico, to derive library fragments from any set of structures.
Decomposition and synthesis
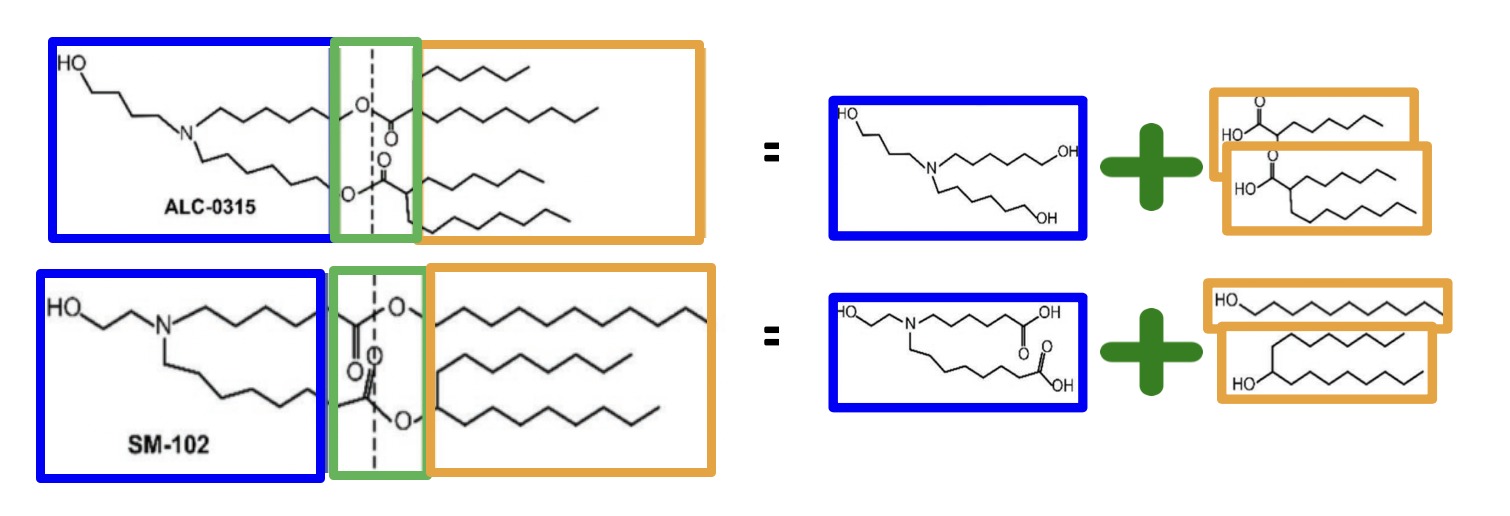
Recall that the renowned ionizable lipids in the Pfizer-BioNTech and Moderna mRNA vaccines for COVID-19, known as ALC-0315 (Pfizer-BioNTech) and SM-102 (Moderna) respectively, have a modular structure, with fragments joined together by (ester) linkages.
(a) Jorgensen et al. 2023
Figure 1: Adapted from Jorgensen et al. 2023.
The combinatorial synthesis of these lipids can be achieved by joining together the fragments – going from right to left in the figure above. In principle, we can also switch the products and reactants, and run the resulting reaction to decompose a structure into its fragments – going from left to right.
Why in silico decomposition?
We’ll implement an in silico structure decomposition method that is general enough to handle a wide variety of structures. This is an extremely useful tool to have, as we will demonstrate below:
Handling the combinatorics: The fragments used to build a structure are the natural way to systematically describe variation in a library. A structure’s fragments determine its composition and therefore its physical properties. Typically, there is tremendous redundancy among the fragments used to be a library of structures. It is therefore often efficient to run various computational methods on fragments individually and put the results together, rather than running them on the structures themselves.
Fragment library generation: The decomposition method can be used to generate a library of fragments from a set of structures, which can then be used for combinatorial synthesis.
Establishing decomposition reactions from synthesis reactions
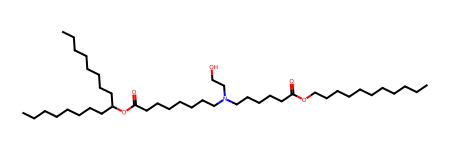
We can test all of this on standard LNP ionizable lipids, such as ALC-0315, which has the following SMILES, retrieved from the known_IL_combined.tsv file written in this post.
Let’s work with just one decomposition reaction for now – the breaking of the ester linkage (saponification) that is reflected in decomposing ALC-0315 and SM-102 as indicated above. The corresponding synthesis reaction is a simple esterification, and can be represented as follows:
How can we use such reactions in silico? First, we recall code from the post on synthesis that is used to run the reaction from a SMARTS specification and reactants.
CODE
from rdkit import Chemfrom rdkit.Chem import AllChem# Flatten the list of lists recursivelydef list_flatten(lst): result = []for item in lst:ifisinstance(item, list): result.extend(list_flatten(item))else: result.append(item)return resultdef run_reaction( smarts_rxn, reactants, one_reaction_only=True):"""Run a chemical reaction on a set of reactants. Parameters ---------- smarts_rxn : :obj:`str` SMARTS string of the reaction. reactants : :obj:`list` of :obj:`str` List of SMILES strings of the reactants. Returns ------- :obj:`list` of :obj:`str` : List of SMILES strings of the products. """ rxn = AllChem.ReactionFromSmarts(smarts_rxn) arglist = []for arg in reactants: arglist.append(Chem.MolFromSmiles(arg)) products = rxn.RunReactants(tuple(arglist)) toret = []for product_set in products: product_list = []for product in product_set: smi = Chem.CanonSmiles( Chem.MolToSmiles(Chem.RemoveHs(product)) ) product_list.append(smi) toret.append(product_list)if one_reaction_only:breakreturn toret
We also need some convenience code to manipulate SMARTS to reverse it.
CODE
def reverse_reaction_from_smarts(smartsin):"""Reverse a chemical reaction. Parameters ---------- smartsin : :obj:`str` SMARTS string of the reaction. Returns ------- :obj:`str` : SMARTS string of the reverse reaction. """ reactants, products = smartsin.split(">>")[0], smartsin.split(">>")[1] smartsout =">>".join([products, reactants])return smartsoutdef reaction_name_to_smarts(reaction_type, rxn_smarts_dict, decompose_mode=False): rxn_smarts = rxn_smarts_dict[reaction_type]if decompose_mode: rxn_smarts = reverse_reaction_from_smarts(rxn_smarts)return rxn_smarts
We can wrap this code to reverse the direction of the reaction if decomposition is being performed.
This code runs the reaction in reverse. The decomposition reaction products are each lighter than the reactants. Therefore, when a decomposition reaction is run repeatedly, it will eventually not have an effect any longer on any of the resulting structural fragments.
So this is our goal in order to decompose a structure with a reaction: run it iteratively until it can run no longer. Here is code to iteratively run such a decomposition reaction on a molecule and record products that result. The decomposition needs to be interleaved with bookkeeping on the reactants, to ensure smooth progress and track all outputs until termination.
CODE
def rxn_decompose_molecules( frag_list, rxn_smarts_dict, reaction_type="esterification", print_debug=False, molecule_info={}, one_reaction_only=True):"""This function uses a chemical reaction to decompose a molecule into fragments. It applies the reaction once to the molecule, collecting all products that result if `one_reaction_only` == False. Parameters ---------- frag_list : :obj:`list` of :obj:`str` List of SMILES strings of the molecules to decompose. reaction_type : :obj:`str`, optional Name of the reaction to use. Default is ``'esterification'``. print_debug : :obj:`bool`, optional If True, print debug statements. Default is False. Returns ------- :obj:`list` of :obj:`str` : List of SMILES strings of product fragments of molecules from frag_list. :obj:`dict` : Decomposition dictionary, mapping each product molecule to a reaction and fragments that constitute it. """ final_frags = []for smiles_to_decompose in frag_list: smiles_to_decompose = Chem.CanonSmiles(smiles_to_decompose)if print_debug:print("Decomposing: ", smiles_to_decompose) products = react_fragments( [smiles_to_decompose], rxn_smarts_dict, reaction_type=reaction_type, decompose_mode=True, one_reaction_only=one_reaction_only )if print_debug:print("Products: ", products)iflen(products) ==0: # This is a fragment - it doesn't decompose# Don't modify molecule_info if no decomposition final_frags.append(smiles_to_decompose)else: chems = list_flatten(products)# We can exhaustively check by actually reacting these reactants.# If we get the same products, then we know that this is a valid decomposition.# If not, then we may need to swap the reactants.iflen(chems) ==1: # Must be duplicated reactants, since this is a binary reaction chems += chems molecule_info[smiles_to_decompose] = {"components": chems,"reaction_type": reaction_type, } final_frags.extend(products) final_frag_list = list_flatten(final_frags) final_frags =list(set(final_frag_list))return final_frags, molecule_info
Typically, there is a list of decomposition reactions given to the method, not just one. This is the topic of another post, which extends the functionality in this one.
It’s instructive to see the results of this decomposition when this is run until termination. For that, we have to wrap it in an iterative loop.
CODE
def decompose_lipids_onestep( init_smiles, rxn_smarts_dict, reaction_types=["esterification"], molecule_info={}, one_reaction_only=True, print_debug=False):"""Decompose a set of molecules into fragments. Parameters ---------- init_smiles : :obj:`list` of :obj:`str` List of SMILES strings of the molecules to decompose. reaction_types : :obj:`list` of :obj:`str`, optional Names of the reactions to use. Default is all reactions. molecule_info : :obj:`dict`, optional Decomposition dictionary, mapping each product molecule to a reaction and fragments that constitute it """ working_frags = init_smiles.copy()for rt in reaction_types: len_rt =0whilelen(working_frags) != len_rt: len_rt =len(working_frags) working_frags, molecule_info = rxn_decompose_molecules( working_frags, rxn_smarts_dict, reaction_type=rt, molecule_info=molecule_info, one_reaction_only=one_reaction_only, print_debug=print_debug )if (len(working_frags) != len_rt) and print_debug:print("Decomposed {} molecules to {} by reversing {} reactions".format( len_rt, len(working_frags), rt ))return working_frags, molecule_infodef full_decompose( frag_list, rxn_smarts_dict, molecule_info={}, reaction_types=["esterification"], one_reaction_only=True, print_debug=False):"""Fully decompose a set of molecules into fragments. Parameters ---------- frag_list : :obj:`list` of :obj:`str` List of SMILES strings of the molecules to decompose. reaction_types : :obj:`list` of :obj:`str`, optional Names of the reactions to use. Default is all reactions. one_reaction_only : :obj:`bool`, optional If True, return only one way of decomposing any reactant. Default is True. Returns ------- :obj:`list` of :obj:`str` : List of SMILES strings of product fragments of molecules from frag_list. :obj:`dict` : Decomposition dictionary, mapping each product molecule to a reaction and fragments that constitute it """ oldlen =0 i =0whilelen(frag_list) != oldlen: oldlen =len(frag_list) frag_list, molecule_info = decompose_lipids_onestep( frag_list, rxn_smarts_dict, reaction_types=reaction_types, molecule_info=molecule_info, one_reaction_only=one_reaction_only, print_debug=print_debug, ) i +=1return frag_list, molecule_info
As we see above, this function full_decompose returns two outputs which are useful for bookkeeping:
A list of the fragments that were generated from the decomposition.
A dictionary which maps each product to the reactants that were used to generate it.
CODE
import numpy as np, pandas as pd, mols2gridlist_prods, molecule_info = full_decompose([smiles_sm102], rxn_smarts_dict, print_debug=True, reaction_types=["esterification"], one_reaction_only=True)mols2grid.display([Chem.MolFromSmiles(x) for x in list_prods],mol_col="mol", n_cols=7, n_rows=4)# rxn_decompose_molecules(list_prods, print_debug=True, reaction_type="esterification", one_reaction_only=True)
Consider a fairly comprehensive set of the known ILs of commercial relevance in drug development. This set of structures, comprising the majority of known named LNPs, is procured from purchasable LNP catalogs.
This is a set of >500 structures comprising ionizable lipid leads and lead candidates in various research programs. We can now decompose all these molecules jointly at all their ester linkages and examine the fragments that result. Notably, very few distinct fragments are required to describe these ionizable lipids.
We will show this now along with an implementation. To set this up, some utility code processes the molecule info dictionary to determine the fragment composition of any of the input structures. The first step to doing this is simply to plot how many fragments the structures have by this decomposition. This is a reflection of the number of ester linkages in the molecule, because every one is cleaved by this in silico reaction.
CODE
def decompose_into_smiles(smiles, all_molecule_info):# Returns a list of fragments comprising the molecule.if smiles notin all_molecule_info:return [smiles] components = all_molecule_info[smiles]['components'] return_list = []for component in components: return_list.extend(decompose_into_smiles(component, all_molecule_info))return return_list
CODE
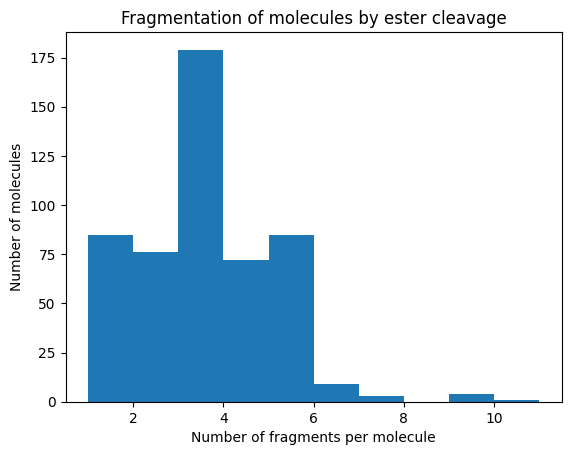
all_smiles =list(all_structures['SMILES'])all_fragments, all_molecule_info = full_decompose( all_smiles, rxn_smarts_dict, reaction_types=["esterification"], one_reaction_only=True)print("{} total fragments represent {} structures: ".format(len(all_fragments), len(all_smiles)))import matplotlib.pyplot as pltplt.hist([len(decompose_into_smiles(x, all_molecule_info)) for x in all_smiles])plt.xlabel("Number of fragments per molecule")plt.ylabel("Number of molecules")plt.title("Fragmentation of molecules by ester cleavage")plt.show()
429 total fragments represent 557 structures:
We see that some structures are not decomposable in this manner because they lack ester linkages, but these constitute <15% of the structures.
The rest have more fragments, with the mode being structures with 3 fragments. By itself, this attests to the structure of the chemical space explored so far. In this manner, we can almost universally express the modular structure of ionizable lipids.
Representing a non-unique decomposition
Each decomposition of a structure defines a tree-structured fragment-based representation of it. The decomposition tree is a non-unique representation of the structure, as there are many ways to decompose a structure into its fragments. But it can be converted into other representations that allow us to visualize the fragments and their relationships.
A small introductory example is representing a structure as an infix string – in essence, an arithmetic expression where the operations are reactions and the operands are structures, which is read in a certain manner and evaluated to give a structure.
This is universal – any such decomposition tree is in the form of a parse tree – its non-leaf nodes are reactions, and its leaf nodes are fragments – which can be converted into an infix string. It is also convenient, converting a SMILES string into a much more human-readable string of a similar length.
Structure → infix string
It’s straightforward to view infix strings like arithmetic expressions. The value is being operated on, and is a number in arithmetic; in this case, the values are fragments or intermediate structures. The operators, which take in values and output values, are in this case reactions. To simplify operations, we deal with only binary reactions with two inputs here.
Code to serialize a structure to an infix string is below.
CODE
# Serialization code for each reaction type.rxn_codes = {"esterification": "R1",}def generate_infix_smiles_decomp( lpd, molecule_info_program, frags_program, rxn_codes, delimiter="_", parens="()"):""" Generate an infix expression for a molecule, based on a decomposition according to database reactions. Works only on binary reactions at present! Parameters ---------- lpd : :obj:`str` SMILES string of the input molecule. molecule_info_program : :obj:`dict` Decomposition dictionary, mapping each product molecule to a reaction and fragments that constitute it. frags_program : :obj:`list` of :obj:`str` List of SMILES strings of the fragments. delimiter : :obj:`str`, optional Delimiter to use between components. Default is ``'_'``. parens : :obj:`str`, optional Parentheses to use around the molecule. Default is ``'()'``. Returns ------- :obj:`str` : Infix expression for the molecule, containing SMILES of all components. """if lpd notin molecule_info_program: # it's a fragmentreturn lpd components = molecule_info_program[lpd]["components"] reaction_type = molecule_info_program[lpd]["reaction_type"]# Check if this is a single product, repeated twice (e.g. symmetric disulfide formation)if (len(components) ==1 ): # Must be duplicated reactants, since this is a binary reaction components += components component_exprs = [ generate_infix_smiles_decomp(c, molecule_info_program, frags_program, rxn_codes)for c in components ] expr_list = [ parens[0], component_exprs[0], rxn_codes[reaction_type], component_exprs[1], parens[1], ]for i inrange(len(expr_list)):if (expr_list[i] in ["(", ")"]) or expr_list[i].startswith("R"):continueif expr_list[i] in frags_program: frag_ID ="F"+str(frags_program.index(expr_list[i])) expr_list[i] = frag_ID# print('Previously unknown fragment: ' + expr_list[i])returnf"{delimiter}".join(expr_list)
This code produces infix expressions with R1 as the placeholder for the reaction (esterification) and F0, F1, … as the placeholders for the fragments.
CODE
rxn_str = generate_infix_smiles_decomp( smiles_sm102, all_molecule_info, all_fragments, rxn_codes)print("Infix expression for SM-102:\t{}".format(rxn_str))
Infix expression for SM-102: (_(_F174_R1_F301_)_R1_F126_)
Displaying these fragments, we see that they are the same ones obtained in the use of full_decompose above.
CODE
frag_ndces = [int(x[1:]) for x in rxn_str.split("_") if x.startswith("F")]list_prods = np.array(all_fragments)[frag_ndces]mols2grid.display([Chem.MolFromSmiles(x) for x in list_prods],mol_col="mol", n_cols=7, n_rows=4)
Infix string → structures
As I mentioned, parse trees and infix strings are essentially equivalent, and we can convert any infix string to the corresponding SMILES as well with knowledge of the decomposition’s fragment IDs.
The topic of visualizing these types of decompositions is covered in another post, but we include some code for completeness here, to evaluate the infix string to get any resulting SMILES.
CODE
import itertoolsdef evaluate_expression( infix_str, frags_program, rxn_smarts_dict, rxn_codes):""" Returns a list of SMILES strings that are the possible results of evaluating the infix expression infix_str. frags_program is a list of SMILES strings that correspond to fragment IDs in the expression. random_choice is the number of random fragments to subsample and add to the expression (None for no subsampling). """ rxn_names = {v: k for k, v in rxn_codes.items()} tokens = infix_str.split('_')# Base case: if token is a SMILES, return its valueiflen(tokens) ==1:if tokens[0].startswith('F'): # It's a fragment this_frag_smiles = frags_program[int(tokens[0][1:])] toret = [ this_frag_smiles ] # Add the fragment that constituted the original structure possible_results = [Chem.CanonSmiles(x) for x inlist(set(toret))]else: # It's a SMILES string possible_results = [ Chem.CanonSmiles(tokens[0]) ]return possible_results# Handle Parenthesesfor i, ch inenumerate(tokens):if ch =='(': start = ielif ch ==')': end = i mid_values = evaluate_expression('_'.join(tokens[start+1:end]), frags_program, rxn_smarts_dict, rxn_codes) toret = [] leftstr ='_'.join(tokens[:start]) rightstr ='_'.join(tokens[end+1:])for left_val in mid_values: strlst = []iflen(leftstr) >0: strlst.append(leftstr) strlst.append(str(left_val))iflen(rightstr) >0: strlst.append(rightstr) toret.extend(evaluate_expression('_'.join(strlst), frags_program, rxn_smarts_dict, rxn_codes))return toret# Handle reactionsfor i inreversed(range(len(tokens))):if tokens[i].startswith('R'): toret = [] left_vals = evaluate_expression('_'.join(tokens[:i]), frags_program, rxn_smarts_dict, rxn_codes) right_vals = evaluate_expression('_'.join(tokens[i+1:]), frags_program, rxn_smarts_dict, rxn_codes) reaction_code = tokens[i]for comp1, comp2 in itertools.product(left_vals, right_vals): reactants = [comp1, comp2] reaction_name = rxn_names[reaction_code] products = react_fragments( reactants, rxn_smarts_dict, reaction_type=reaction_name, decompose_mode=False, one_reaction_only=False) toret.extend(list(set(list_flatten(products))))return toret
The algorithm to do this is a classic application of stacks devised by the computer scientist Edsger Dijkstra, called the shunting yard algorithm. The algorithm uses two stacks: the reaction stack and the output queue. Reading tokens left‑to‑right, it immediately pushes SMILES fragments/structures onto the output. When it encounters a reaction, it can compare its precedence and associativity with the reaction on the top of the reaction stack, and accordingly pop operators on the stack to the output before the new operator is pushed. Left parentheses are pushed onto the reaction stack to mark sub‑expressions; when a right parenthesis appears, reactions are popped to the output until the matching left parenthesis is removed to finish evaluating the sub-expression. After all tokens are processed, any remaining reactions are popped to the output queue. The resulting postfix sequence can be evaluated in a single pass with one stack, giving the final SMILES result.
From synthesis to decomposition and back again
Now that we’ve set up synthesis and decomposition routines, it’s interesting to use them together in larger pipelines. But this is not straightforward. Decomposition is not unique – there are many ways to decompose a structure, corresponding to different orders of peeling off the fragments. The same is true for synthesis, with reactive sites not being unique either.
Decomposition and synthesis: in silico inverses of each other
For our purposes, the in silico decomposition reaction should be reversible. This means that the decomposition reaction should be possible to run in reverse, and the products should be possible to reassemble into the original structure. Otherwise, the decomposition into fragments loses valuable information about the structure.
We can check that when we take a structure and decompose it into fragments, we can run these fragments through the appropriate synthesis routines and recover the structure. This is useful to check for in general, and we start with the already-decomposed structure of SM-102.
CODE
lps =set([Chem.CanonSmiles(x) for x in evaluate_expression(rxn_str, all_fragments, rxn_smarts_dict, rxn_codes)])mols2grid.display([Chem.MolFromSmiles(x) for x in lps], n_cols=7, n_rows=4)
CODE
lps2 =set([Chem.CanonSmiles(x) for x in evaluate_expression(rxn_str, all_fragments, rxn_smarts_dict, rxn_codes)])mols2grid.display([Chem.MolFromSmiles(x) for x in lps2], n_cols=7, n_rows=4)
There are two structures instead of one when we reconstitute the fragments. It’s worth understanding why – this is because they can react on either of the two carboxylic acid groups on the head, which is asymmetric. Crucially, SM-102 itself is in the reconstituted set:
CODE
print(smiles_sm102 in lps)
True
This is a key desirable property to keep in mind when we start coupling decomposition and synthesis to create combinatorially varied virtual spaces:
When we decompose a structure and reconstitute the fragments (with the same reaction settings), we always recover at least the original structure.
In other words, the decomposition and synthesis reactions are in silico inverses of each other.
Decompose all chemicals at once
We can now decompose all these structures at once to get a sense of which fragments they share, and sanity-check our insights so far.
CODE
def decompose_dataset( input_lipids, rxn_smarts_dict, rxn_codes, reaction_types=["esterification"], print_debug=False): input_lipids = [Chem.CanonSmiles(x) for x in input_lipids] frags_program = input_lipids oldlen =0while oldlen !=len(frags_program): oldlen =len(frags_program) frags_program, molecule_info_program = full_decompose( frags_program, rxn_smarts_dict, reaction_types=reaction_types, one_reaction_only=True, print_debug=print_debug, ) input_lipid_desc = {}for lpd in input_lipids: input_lipid_desc[lpd] = generate_infix_smiles_decomp( lpd, molecule_info_program, frags_program, rxn_codes )return input_lipid_desc, molecule_info_program, frags_program# Get the fragment counts for each structure in input_lipid_desc. # This should be a dictionary mapping fragment names to counts in the subsuming structure.def compute_fragment_counts( input_lipid_desc, frags_program, rxn_codes, default_pfx ='all_chems/'): all_frags = [] uq_lipid_frags = [] aggregate_counts = {}for smi in input_lipid_desc: these_counts = np.zeros(len(frags_program)).astype(int) tag = input_lipid_desc[smi] frags = [x for x in tag.split('_') if x.startswith('F')] all_frags += frags uq_lipid_frags +=list(set(frags))for f in frags: frags_program_index =int(f[1:]) these_counts[frags_program_index] +=1 aggregate_counts[smi] = these_counts uaf = np.unique(all_frags, return_counts=True) uaf_dict =dict(zip(uaf[0], uaf[1])) ulf = np.unique(uq_lipid_frags, return_counts=True) ulf_dict =dict(zip(ulf[0], ulf[1])) name_arr = ['F'+str(x) for x inrange(len(frags_program))] lipid_tags_df = pd.DataFrame.from_dict({'SMILES': input_lipid_desc.keys(), 'Tag': input_lipid_desc.values()}) rxn_codes_df = pd.DataFrame.from_dict({'Reaction Name': rxn_codes.keys(), 'Reaction code': rxn_codes.values()})#print(uaf_dict) frag_count_df = pd.DataFrame.from_dict({'Name': name_arr, 'SMILES': frags_program, 'Count_in_library': [uaf_dict.get(x, 0) for x in name_arr], 'Number_of_unique_lipids': [ulf_dict.get(x, 0) for x in name_arr] }) frag_count_df = pd.concat([frag_count_df, pd.DataFrame.from_dict(aggregate_counts)], axis=1)if default_pfx isnotNone:ifnot os.path.exists(default_pfx): os.makedirs(default_pfx) lipid_tags_df.to_csv(f'{default_pfx}lipid_tags.tsv', sep='\t', index=False) rxn_codes_df.to_csv(f'{default_pfx}rxn_codes.tsv', sep='\t', index=False) frag_count_df.to_csv(f'{default_pfx}fragments.tsv', sep='\t', index=False)return lipid_tags_df, frag_count_df, rxn_codes_df
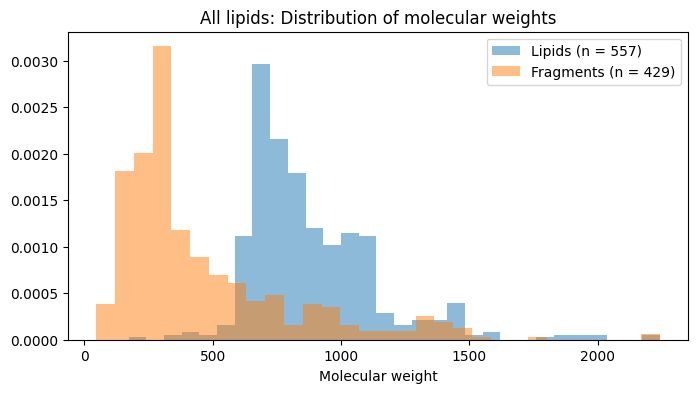
How much are we gaining by decomposing structures in this straightforward manner? It depends on the set of structures and how much redundancy they share. For a varied set such as those in the catalogs here which are discovered through completely different pipelines and routes, the amount of redundancy we would expect is not high.
It is surprising therefore that the decompositions are nontrivial, as we saw with most structures being cleaved. This results in significantly lighter (less complex) fragments, a fact which can be easily plotted.
CODE
import matplotlib.pyplot as pltinput_lipid_desc, molecule_info_program, frags_program = decompose_dataset(all_smiles, rxn_smarts_dict, rxn_codes, reaction_types=['esterification'])lipid_tags_df, frag_count_df, rxn_codes_df = compute_fragment_counts(input_lipid_desc, frags_program, rxn_codes)frag_count_df.set_index('SMILES', inplace=True)newcols =list(frag_count_df.columns)for i inrange(3, frag_count_df.shape[1]): sndx = np.where(np.array(all_smiles) == frag_count_df.columns[i])[0]iflen(sndx) >0: newcols[i] = all_structures['name'][sndx[0]]frag_count_df.columns = newcolsnewrows =list(frag_count_df.index)for i inrange(frag_count_df.shape[0]): sndx = np.where(np.array(all_smiles) == frag_count_df.columns[i])[0]iflen(sndx) >0: newcols[i] = all_structures['name'][sndx[0]]frag_count_df.index = newrowsimport rdkit.Chem.Descriptorsdata_chemwts = [Chem.Descriptors.ExactMolWt(Chem.MolFromSmiles(x)) for x in all_smiles]data_fragwts = [Chem.Descriptors.ExactMolWt(Chem.MolFromSmiles(x)) for x in frags_program]data1 = data_chemwts + data_fragwtslbls1 = ['structures']*len(data_chemwts) + ['fragments']*len(data_fragwts)def plot_hist_molwts(data1, data2, fam_name): plt.figure(figsize=(8, 4)) plt.hist(data1, bins=30, label=f'Lipids (n = {len(data1)})', alpha=0.5, density=True) plt.hist(data2, bins=30, label=f'Fragments (n = {len(data2)})', alpha=0.5, density=True) plt.xlabel("Molecular weight") plt.title(f"{fam_name}: Distribution of molecular weights") plt.legend() plt.show()plot_hist_molwts(data_chemwts, data_fragwts, 'All lipids')
Now we can further analyze the table of the (discrete) fragment count of each of the lipid structures. The most frequently occurring fragments are a heterogeneous mix.
CODE
sorted_frag_df = frag_count_df.iloc[np.argsort(frag_count_df['Number_of_unique_lipids'])[::-1], :]mols2grid.display( pd.DataFrame({'SMILES': list(sorted_frag_df.index), 'numuq': ["In {} unique lipids".format(x) for x in sorted_frag_df['Number_of_unique_lipids']] }), subset=['img', 'numuq'], n_cols=7, n_rows=4)
The most frequently occurring fragments are a mix of the tertiary amine heads we are accustomed to, along with generic medium-length hydrophobic tail fragments, mostly unsaturated with a small amount of branching.
Unfortunately, it is also clear that there are still a few very heavy fragments, resulting from lipids that are not cleaved:
CODE
from rdkit.Chem import Descriptorsdesc_molwt_ndces = np.argsort([Descriptors.MolWt(Chem.MolFromSmiles(x)) for x in frags_program])[::-1]mols2grid.display( pd.DataFrame({'SMILES': np.array(frags_program)[desc_molwt_ndces], 'numuq': np.array(["{:.2f} Da".format(Descriptors.MolWt(Chem.MolFromSmiles(x))) for x in frags_program])[desc_molwt_ndces] }), subset=['img', 'numuq'], title="Fragments sorted by molecular weight (descending order)")
The first few dozen of these fragments consist of many huge structures that must each be painstakingly constructed, and therefore are appropriately viewed as compositions of fragments themselves, through other non-esterification reactions.
In order to decompose using multiple reactions at once, there is a little further development required, for which we refer to another post.
Exporting code
For future such reference, it’s useful to collect all the code from this post in a .py file.
CODE
all_code =r'''from rdkit import Chemfrom rdkit.Chem import AllChemimport random, itertoolsimport numpy as np, pandas as pdfrom rdkit.Chem.Draw import rdMolDraw2Dfrom rdkit.Chem import rdChemReactionsimport io, osfrom PIL import Imagedef draw_reaction(smarts_in, width=700, height=300): rxn = rdChemReactions.ReactionFromSmarts(smarts_in) drawer = rdMolDraw2D.MolDraw2DCairo(width, height) drawer.DrawReaction(rxn) drawer.FinishDrawing() return drawer.GetDrawingText()def reverse_reaction_from_smarts(smartsin): """Reverse a chemical reaction. Parameters ---------- smartsin : :obj:`str` SMARTS string of the reaction. Returns ------- :obj:`str` : SMARTS string of the reverse reaction. """ reactants, products = smartsin.split(">>")[0], smartsin.split(">>")[1] smartsout = ">>".join([products, reactants]) return smartsoutdef reaction_name_to_smarts(reaction_type, rxn_smarts_dict, decompose_mode=False): rxn_smarts = rxn_smarts_dict[reaction_type] if decompose_mode: rxn_smarts = reverse_reaction_from_smarts(rxn_smarts) return rxn_smarts# Flatten the list of lists recursivelydef list_flatten(lst): result = [] for item in lst: if isinstance(item, list): result.extend(list_flatten(item)) else: result.append(item) return resultdef run_reaction( smarts_rxn, reactants, one_reaction_only=True): """Run a chemical reaction on a set of reactants. Parameters ---------- smarts_rxn : :obj:`str` SMARTS string of the reaction. reactants : :obj:`list` of :obj:`str` List of SMILES strings of the reactants. Returns ------- :obj:`list` of :obj:`str` : List of SMILES strings of the products. """ rxn = AllChem.ReactionFromSmarts(smarts_rxn) arglist = [] for arg in reactants: arglist.append(Chem.MolFromSmiles(arg)) products = rxn.RunReactants(tuple(arglist)) toret = [] for product_set in products: product_list = [] for product in product_set: smi = Chem.CanonSmiles( Chem.MolToSmiles(Chem.RemoveHs(product))) product_list.append(smi) toret.append(product_list) if one_reaction_only: break return toretdef react_fragments( reactants, rxn_smarts_dict, reaction_type="esterification", decompose_mode=False, one_reaction_only=True): rxn_smarts = reaction_name_to_smarts(reaction_type, rxn_smarts_dict, decompose_mode=decompose_mode) if rxn_smarts == "": return [] else: return run_reaction(rxn_smarts, reactants, one_reaction_only=one_reaction_only)def rxn_decompose_molecules( frag_list, rxn_smarts_dict, reaction_type="esterification", print_debug=False, molecule_info={}, one_reaction_only=True): """This function uses a chemical reaction to decompose a molecule into fragments. It applies the reaction once to the molecule, collecting all products that result if `one_reaction_only` == False. Parameters ---------- frag_list : :obj:`list` of :obj:`str` List of SMILES strings of the molecules to decompose. reaction_type : :obj:`str`, optional Name of the reaction to use. Default is ``'esterification'``. print_debug : :obj:`bool`, optional If True, print debug statements. Default is False. Returns ------- :obj:`list` of :obj:`str` : List of SMILES strings of product fragments of molecules from frag_list. :obj:`dict` : Decomposition dictionary, mapping each product molecule to a reaction and fragments that constitute it. """ final_frags = [] for smiles_to_decompose in frag_list: smiles_to_decompose = Chem.CanonSmiles(smiles_to_decompose) if print_debug: print("Decomposing: ", smiles_to_decompose) products = react_fragments([smiles_to_decompose], rxn_smarts_dict, reaction_type=reaction_type, decompose_mode=True, one_reaction_only=one_reaction_only) if print_debug: print("Products: ", products) if len(products) == 0: # This is a fragment - it doesn't decompose# Don't modify molecule_info if no decomposition final_frags.append(smiles_to_decompose) else: chems = list_flatten(products)# We can exhaustively check by actually reacting these reactants.# If we get the same products, then we know that this is a valid decomposition.# If not, then we may need to swap the reactants. if len(chems) == 1: # Must be duplicated reactants, since this is a binary reaction chems += chems molecule_info[smiles_to_decompose] = { "components": chems, "reaction_type": reaction_type, } final_frags.extend(products) final_frag_list = list_flatten(final_frags) final_frags = list(set(final_frag_list)) return final_frags, molecule_infodef decompose_lipids_onestep( init_smiles, rxn_smarts_dict, reaction_types=["esterification"], molecule_info={}, one_reaction_only=True, print_debug=False): """Decompose a set of molecules into fragments. Parameters ---------- init_smiles : :obj:`list` of :obj:`str` List of SMILES strings of the molecules to decompose. reaction_types : :obj:`list` of :obj:`str`, optional Names of the reactions to use. Default is all reactions. molecule_info : :obj:`dict`, optional Decomposition dictionary, mapping each product molecule to a reaction and fragments that constitute it """ working_frags = init_smiles.copy() for rt in reaction_types: len_rt = 0 while len(working_frags) != len_rt: len_rt = len(working_frags) working_frags, molecule_info = rxn_decompose_molecules( working_frags, rxn_smarts_dict, reaction_type=rt, molecule_info=molecule_info, one_reaction_only=one_reaction_only, print_debug=print_debug) if (len(working_frags) != len_rt) and print_debug: print( "Decomposed {} molecules to {} by reversing {} reactions".format( len_rt, len(working_frags), rt)) return working_frags, molecule_infodef full_decompose( frag_list, rxn_smarts_dict, molecule_info={}, reaction_types=["esterification"], one_reaction_only=True, print_debug=False): """Fully decompose a set of molecules into fragments. Parameters ---------- frag_list : :obj:`list` of :obj:`str` List of SMILES strings of the molecules to decompose. reaction_types : :obj:`list` of :obj:`str`, optional Names of the reactions to use. Default is all reactions. one_reaction_only : :obj:`bool`, optional If True, return only one way of decomposing any reactant. Default is True. Returns ------- :obj:`list` of :obj:`str` : List of SMILES strings of product fragments of molecules from frag_list. :obj:`dict` : Decomposition dictionary, mapping each product molecule to a reaction and fragments that constitute it """ oldlen = 0 i = 0 while len(frag_list) != oldlen: oldlen = len(frag_list) frag_list, molecule_info = decompose_lipids_onestep( frag_list, rxn_smarts_dict, reaction_types=reaction_types, molecule_info=molecule_info, one_reaction_only=one_reaction_only, print_debug=print_debug,) i += 1 return frag_list, molecule_infodef decompose_into_smiles(smiles, all_molecule_info):# Returns a list of fragments comprising the molecule. if smiles not in all_molecule_info: return [smiles] components = all_molecule_info[smiles]['components'] return_list = [] for component in components: return_list.extend(decompose_into_smiles(component, all_molecule_info)) return return_listdef generate_infix_smiles_decomp( lpd, molecule_info_program, frags_program, rxn_codes, delimiter="_", parens="()"): """ Generate an infix expression for a molecule, based on a decomposition according to database reactions. Works only on binary reactions at present! Parameters ---------- lpd : :obj:`str` SMILES string of the input molecule. molecule_info_program : :obj:`dict` Decomposition dictionary, mapping each product molecule to a reaction and fragments that constitute it. frags_program : :obj:`list` of :obj:`str` List of SMILES strings of the fragments. delimiter : :obj:`str`, optional Delimiter to use between components. Default is ``'_'``. parens : :obj:`str`, optional Parentheses to use around the molecule. Default is ``'()'``. Returns ------- :obj:`str` : Infix expression for the molecule, containing SMILES of all components. """ if lpd not in molecule_info_program: # it's a fragment return lpd components = molecule_info_program[lpd]["components"] reaction_type = molecule_info_program[lpd]["reaction_type"]# Check if this is a single product, repeated twice (e.g. symmetric disulfide formation) if ( len(components) == 1): # Must be duplicated reactants, since this is a binary reaction components += components component_exprs = [ generate_infix_smiles_decomp(c, molecule_info_program, frags_program, rxn_codes) for c in components ] expr_list = [ parens[0], component_exprs[0], rxn_codes[reaction_type], component_exprs[1], parens[1], ] for i in range(len(expr_list)): if (expr_list[i] in ["(", ")"]) or expr_list[i].startswith("R"): continue if expr_list[i] in frags_program: frag_ID = "F" + str(frags_program.index(expr_list[i])) expr_list[i] = frag_ID# print('Previously unknown fragment: ' + expr_list[i]) return f"{delimiter}".join(expr_list)import itertoolsdef evaluate_expression( infix_str, frags_program, rxn_smarts_dict, rxn_codes): """ Returns a list of SMILES strings that are the possible results of evaluating the infix expression infix_str. frags_program is a list of SMILES strings that correspond to fragment IDs in the expression. random_choice is the number of random fragments to subsample and add to the expression (None for no subsampling). """ rxn_names = {v: k for k, v in rxn_codes.items()} tokens = infix_str.split('_')# Base case: if token is a SMILES, return its value if len(tokens) == 1: if tokens[0].startswith('F'): # It's a fragment this_frag_smiles = frags_program[int(tokens[0][1:])] toret = [ this_frag_smiles ]# Add the fragment that constituted the original structure possible_results = [Chem.CanonSmiles(x) for x in list(set(toret))] else: # It's a SMILES string possible_results = [ Chem.CanonSmiles(tokens[0]) ] return possible_results# Handle Parentheses for i, ch in enumerate(tokens): if ch == '(': start = i elif ch == ')': end = i mid_values = evaluate_expression( '_'.join(tokens[start+1:end]), frags_program, rxn_smarts_dict, rxn_codes) toret = [] leftstr = '_'.join(tokens[:start]) rightstr = '_'.join(tokens[end+1:]) for left_val in mid_values: strlst = [] if len(leftstr) > 0: strlst.append(leftstr) strlst.append(str(left_val)) if len(rightstr) > 0: strlst.append(rightstr) toret.extend(evaluate_expression( '_'.join(strlst), frags_program, rxn_smarts_dict, rxn_codes)) return toret # Handle reactions for i in reversed(range(len(tokens))): if tokens[i].startswith('R'): toret = [] left_vals = evaluate_expression( '_'.join(tokens[:i]), frags_program, rxn_smarts_dict, rxn_codes) right_vals = evaluate_expression( '_'.join(tokens[i+1:]), frags_program, rxn_smarts_dict, rxn_codes) reaction_code = tokens[i] for comp1, comp2 in itertools.product(left_vals, right_vals): reactants = [comp1, comp2] reaction_name = rxn_names[reaction_code] products = react_fragments( reactants, rxn_smarts_dict, reaction_type=reaction_name, decompose_mode=False, one_reaction_only=False) toret.extend(list(set(list_flatten(products)))) return toretdef decompose_dataset( input_lipids, rxn_smarts_dict, rxn_codes, reaction_types=["esterification"], print_debug=False): input_lipids = [Chem.CanonSmiles(x) for x in input_lipids] frags_program = input_lipids oldlen = 0 while oldlen != len(frags_program): oldlen = len(frags_program) frags_program, molecule_info_program = full_decompose( frags_program, rxn_smarts_dict, reaction_types=reaction_types, one_reaction_only=True, print_debug=print_debug,) input_lipid_desc = {} for lpd in input_lipids: input_lipid_desc[lpd] = generate_infix_smiles_decomp( lpd, molecule_info_program, frags_program, rxn_codes) return input_lipid_desc, molecule_info_program, frags_program# Get the fragment counts for each lipid in input_lipid_desc. This should be a dictionary mapping fragment names to counts in the lipid.def compute_fragment_counts( input_lipid_desc, frags_program, rxn_codes, default_pfx = 'all_chems/'): all_frags = [] uq_lipid_frags = [] aggregate_counts = {} for smi in input_lipid_desc: these_counts = np.zeros(len(frags_program)).astype(int) tag = input_lipid_desc[smi] frags = [x for x in tag.split('_') if x.startswith('F')] all_frags += frags uq_lipid_frags += list(set(frags)) for f in frags: frags_program_index = int(f[1:]) these_counts[frags_program_index]+= 1 aggregate_counts[smi] = these_counts uaf = np.unique(all_frags, return_counts=True) uaf_dict = dict(zip(uaf[0], uaf[1])) ulf = np.unique(uq_lipid_frags, return_counts=True) ulf_dict = dict(zip(ulf[0], ulf[1])) name_arr = ['F' + str(x) for x in range(len(frags_program))] lipid_tags_df = pd.DataFrame.from_dict({'SMILES': input_lipid_desc.keys(), 'Tag': input_lipid_desc.values()}) rxn_codes_df = pd.DataFrame.from_dict({'Reaction Name': rxn_codes.keys(), 'Reaction code': rxn_codes.values()})#print(uaf_dict) frag_count_df = pd.DataFrame.from_dict({ 'Name': name_arr, 'SMILES': frags_program, 'Count_in_library': [uaf_dict.get(x, 0) for x in name_arr], 'Number_of_unique_lipids': [ulf_dict.get(x, 0) for x in name_arr] }) frag_count_df = pd.concat([frag_count_df, pd.DataFrame.from_dict(aggregate_counts)], axis=1) if default_pfx is not None: if not os.path.exists(default_pfx): os.makedirs(default_pfx) lipid_tags_df.to_csv(f'{default_pfx}lipid_tags.tsv', sep='\t', index=False) rxn_codes_df.to_csv(f'{default_pfx}rxn_codes.tsv', sep='\t', index=False) frag_count_df.to_csv(f'{default_pfx}fragments.tsv', sep='\t', index=False) return lipid_tags_df, frag_count_df, rxn_codes_df'''
CODE
file_pfx ="../../files/chem/"withopen(file_pfx +"combinatorial_chemistry_decomposition_tools.py", "w", encoding="utf-8") as f: f.write(all_code)
This can be developed in several advanced directions, which are the subject of separate posts.