CODE
file_pfx = "../../files/chem/"
decomp_tools_path = file_pfx + "combinatorial_chemistry_decomposition_tools.py"
from importlib.machinery import SourceFileLoader
ccd_tools = SourceFileLoader("ccd_tools", decomp_tools_path).load_module()This post follows up on a predecessor post which sets up how to decompose chemical structures in silico into fragments. That post shows how a single common fragment composition reaction (esterification) can be viewed in reverse as a cleavage reaction of ester linkages (“saponification”), and in this manner can be repeatedly applied to ionizable lipids in the literature to yield a common set of building block fragments.
Here we focus on expanding this idea to more reactions beyond esterification, giving a pragmatic and general implementation to iteratively run a list of decomposition reactions on the input structure, with a priority order determined by the list.
To demonstrate such functionality, we put together a set of reactions used in ionizable lipid design for LNPs. These reactions collectively form a fairly comprehensive set for:
Our focus is not on being exhaustive, but rather on covering maximal ground with a small set of reactions within IL design, a paradigmatic application of these ultralarge chemical spaces.
We pick up where the predecessor post left off. The first step is to recall the predecessor post’s code, which was saved to a .py file at the end of that post. We can import this code from the assumed file name combinatorial_chemistry_decomposition_tools.py. For explanations and documentation, refer to that post.
file_pfx = "../../files/chem/"
decomp_tools_path = file_pfx + "combinatorial_chemistry_decomposition_tools.py"
from importlib.machinery import SourceFileLoader
ccd_tools = SourceFileLoader("ccd_tools", decomp_tools_path).load_module()The esterification/saponification example outlined in the other post shows clearly how a given reaction principle can be applied in reverse to decompose a molecule into fragments, which can be reconstituted for even greater variety.
from rdkit import Chem
from PIL import Image
import io, os
rxn_smarts_dict = {}
rxn_smarts_dict["esterification"] = (
"[#6:4][C:2](=[O:3])[OX2H1:1]."
"[#6X4,c;!$(C([OX2H])[O,S,#7,#15]):6][OX2H1]"
">>"
"[#6:6][O:1][C:2](=[O:3])[#6:4]"
)
bio = io.BytesIO(ccd_tools.draw_reaction(rxn_smarts_dict["esterification"]))
Image.open(bio)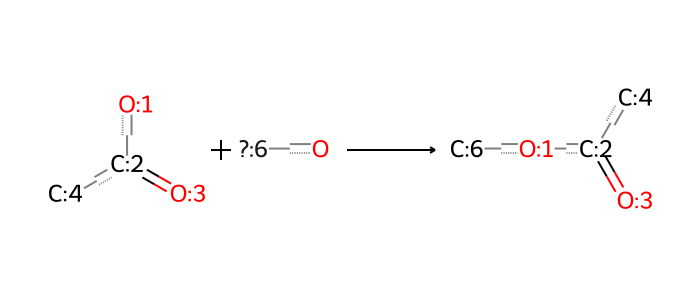
We can apply this same principle to a variety of reactions, and this post compiles a set of reactions that are relevant to ionizable lipid development.
We’ll implement the following steps:
In defining these reactions, a couple of primary goals keep recurring:
Composability: Retaining the ability to compose them, or perform them in repeated sequence. The consequences of this are widely appreciated in in silico synthesis – the reactions should be defined in a way that allows for the products of one reaction to be used as the reactants for another reaction. But for in silico decomposition, too, composability is powerful and relevant, because it’s easy to see when a sequence of decompositions has gone “far enough.”
Reversibility: A synthesis reaction reversed implies a decomposition rule, and vice versa. This is developed formally in silico in the predecessor post – it means that reaction definitions with highly specific products imply highly specific reactants, and vice versa.
This post is one good starting point that addresses these goals, showing how to use rules like those in (Hartenfeller et al. 2011). But typical industrial applications benefit from refining these reaction rules by defining a larger number of more specific reactions, to gain granular control over the fragments that are generated.
We see that when we change the reaction(s) used to decompose a structure, a different set of fragments will result. The idea of a “fragment” is itself contextual, and depends on the beholder; fragments can be heavy or light in different situations, based on the base unit of synthesis.
Our work here gives this an explicit operational definition:
A fragment is “that which cannot be decomposed further” in the context of a given set of reactions. This can only be defined in the context of a set of reactions applied to a set of structures.
This is a powerful way to view fragments and ultralarge spaces, enabling us to freely mine fragments from structures in the literature by in silico decomposition.
In silico combination of chemical fragments is an idealization of a reaction that happens with perfect yield. This is never really achieved under any physical conditions, let alone in industrial practice. But modeling and screening are concerned not with failed syntheses, but with stable candidates that are the intended products of these reactions as written.
For the purposes of defining a virtual space for screening, it’s normally considered appropriate to ignore the practical limitations of chemical synthesis, and instead focus on the idealized reactions that can be used to generate a combinatorial library of fragments. The resulting virtual space will anyway be filtered for stable and effective candidates on the back end, even if not on the front end.
The reactions here are defined rather non-specifically, often not requiring more from a reactant than a single reactive group. In particular design efforts, it is often useful to sharpen the reaction definition (make it more specific) by requiring more precise structural patterns to be present in the reactants. Therefore, the reaction list is not intended to be exhaustive, but it does yield a sensible set of fragments when applied to known ionizable lipids, as seen below.
This principle is useful for any set of reactions. Most ultralarge combinatorial chemical spaces are comprised of relatively few reactions, despite the combinatorics. Even the largest on-demand catalogs of commercially available small drug-like molecules, from vendors like Enamine or WuXi, are based on <200 core reaction rules.
For a specialized set of structures such as ionizable lipids, the number of reactions needed is much smaller. Even just the single reaction of ester linkage formation/cleavage is enough to dramatically decompose most known ionizable lipids.
So we add more reactions that are relevant to ionizable lipid design. A lot of our functionality benefits from thinking of reactions as having two reactants. This is a simplified case but still a general one, as any multi-component reaction can be broken into a series of two-component reactions.
A very useful reaction is amide formation using a primary amine with an acid to make an amide. In our framework, including this implies the reverse amide cleavage reaction when decomposing structures. This is another useful way to get derive primary amine fragments for further screening purposes.
rxn_smarts_dict["amide_coupling"] = (
"[#6:1][C:2](=[O:3])[OX2H1]."
"[#6:6][NX3;H2;!$(NC=O):5]"
">>"
"[#6:6][NX3R0;H1:5][CR0:2](=[O:3])[#6:1]"
)
bio = io.BytesIO(ccd_tools.draw_reaction(rxn_smarts_dict["amide_coupling"]))
Image.open(bio)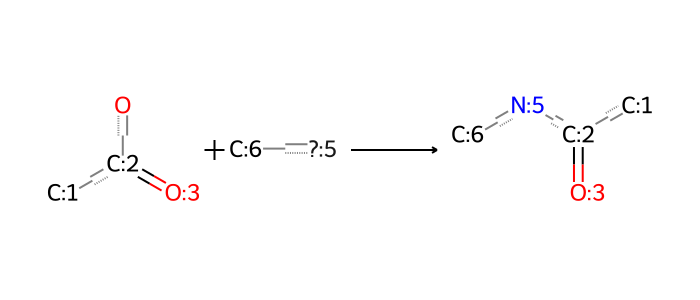
By the same reasoning, we note other reactions that involve amines. A notable one in many papers in the literature is a Michael addition, where a nucleophilic amine attacks an electrophilic alkene. This has proved to be a useful reaction in IL design because it selectively introduces the amine into the structure at a prescribed position relative to the carbonyl carbon (e.g. the \(\gamma\) position here).
For our purposes, the Michael addition has to be defined carefully and somewhat specifically for a variety of reasons, including reversibility and composability. Different versions of the reaction must be defined with primary and secondary amines as the respective nucleophiles. Here is an example with the electrophile being an acrylate, a common Michael acceptor in ILs.
rxn_smarts_dict["michael_addition_acrylate_1o"] = (
"[CX3H2:1]=[CH1:2][C:3](=[O:4])[O:5]."
"[NX3;H2:6]"
">>"
"[NX3;H1:6][CH2:1][CH2:2][C:3](=[O:4])[O:5]"
)
rxn_smarts_dict["michael_addition_acrylate_2o"] = (
"[CX3H2:1]=[CH1:2][C:3](=[O:4])[O:5]."
"[NX3;H1:6]"
">>"
"[NX3;H0:6][CH2:1][CH2:2][C:3](=[O:4])[O:5]"
)
bio = io.BytesIO(ccd_tools.draw_reaction(rxn_smarts_dict["michael_addition_acrylate_1o"]))
Image.open(bio)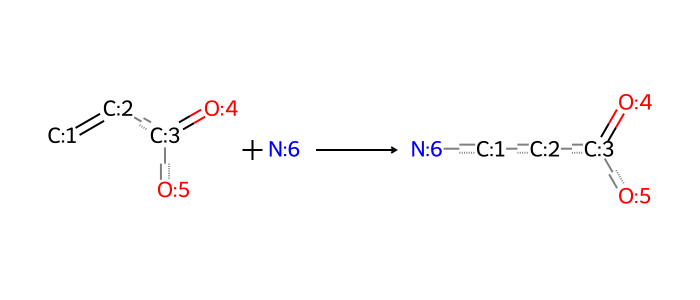
The electrophile is a \((\alpha, \beta)\)-unsaturated carbonyl compound, which is a common motif in many ionizable lipids, but this too leads to separately denoted consequences for acrylates on one hand, and other compounds like acrylamides on the other. So it’s actually more convenient to define the reaction separately for acrylates and acrylamides. The acrylamide reactions are below.
rxn_smarts_dict["michael_addition_acrylamide_1o"] = (
"[CX3H2:1]=[CH1:2][C:3](=[O:4])[N:5]."
"[NX3;H2:6]"
">>"
"[NX3;H1:6][CH2:1][CH2:2][C:3](=[O:4])[N:5]"
)
rxn_smarts_dict["michael_addition_acrylamide_2o"] = (
"[CX3H2:1]=[CH1:2][C:3](=[O:4])[N:5]."
"[NX3;H1:6]"
">>"
"[NX3;H0:6][CH2:1][CH2:2][C:3](=[O:4])[N:5]"
)
bio = io.BytesIO(ccd_tools.draw_reaction(rxn_smarts_dict["michael_addition_acrylamide_1o"]))
Image.open(bio)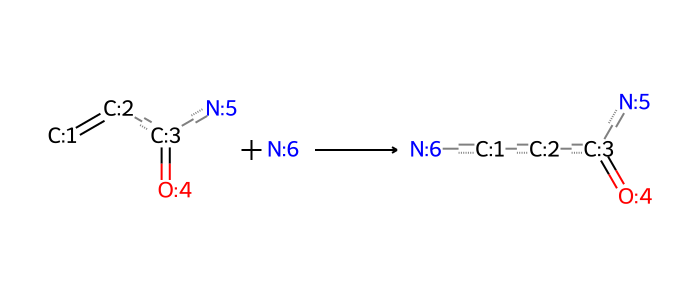
Another set of reactions involving amines in IL chemistry is the attachment of epoxide tails to amines. Here’s how it looks. Again, different rules apply when the nucleophile is a primary or secondary amine, so we define these separately.
rxn_smarts_dict["epoxide_ring_opening_1o"] = (
"[#6:1][CR1:4]1[CR1:3][OR1:2]1."
"[#6:6][NX3;H2;!$(NC=O):5]"
">>"
"[#6:1][CR0:4]([OX2H1:2])[CR0:3][NX3;H1;!$(NC=O):5][#6:6]"
)
rxn_smarts_dict["epoxide_ring_opening_2o"] = (
"[#6:1][CR1:4]1[CR1:3][OR1:2]1."
"[#6:6][NX3;H1;!$(NC=O):5]"
">>"
"[#6:1][CR0:4]([OX2H1:2])[CR0:3][NX3;H0;!$(NC=O):5][#6:6]"
)
bio = io.BytesIO(ccd_tools.draw_reaction(rxn_smarts_dict["epoxide_ring_opening_1o"]))
Image.open(bio)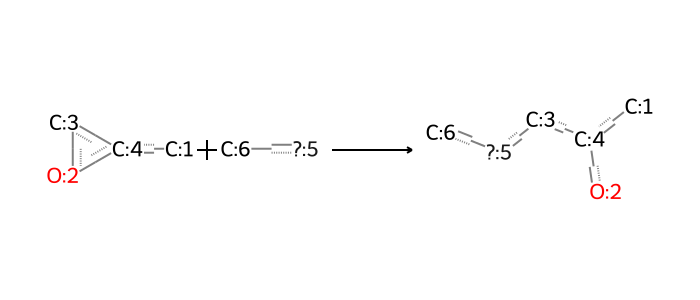
Other relevant reactions here do not involve amines, but can still be useful. Formation of an ether linkage is sometimes used in synthesis of fragments, and can be a useful way to derive common alcohol fragments in a set of structures.
rxn_smarts_dict["alcohols_to_ether"] = (
"[#6:1][OX2H1:2]."
"[#6:3][OX2H1]"
">>"
"[#6:1][OX2H0R0:2][#6:3]"
)
bio = io.BytesIO(ccd_tools.draw_reaction(rxn_smarts_dict["alcohols_to_ether"]))
Image.open(bio)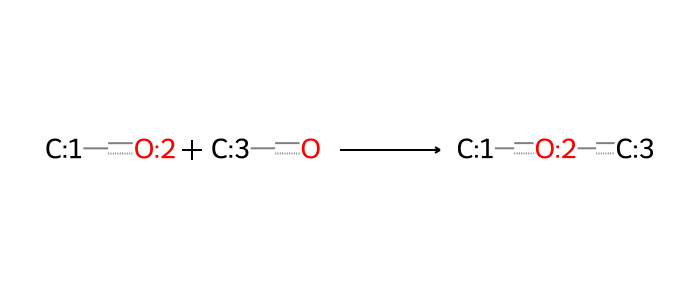
A number of reactions involving sulfur-based linkages are also relevant. The commonest such example involves disulfide bonds as linkages. These are conventionally often thought of as cleavable into thiols, a reaction worth including in efforts where such linkages are relevant. A similar situation arises with thioether linkages, which are also cleavable into thiols.
rxn_smarts_dict["thiol_to_disulfide"] = (
"[SX2H1:1]."
"[SX2H1:2]"
">>"
"[SX2H0:1][SX2H0:2]"
)
rxn_smarts_dict["thiols_to_thioether"] = (
"[#6:1][SX2H1:2]."
"[#6:3][SX2H1]"
">>"
"[#6:1][SX2H0:2][#6:3]"
)
bio = io.BytesIO(ccd_tools.draw_reaction(rxn_smarts_dict["thiols_to_thioether"]))
Image.open(bio)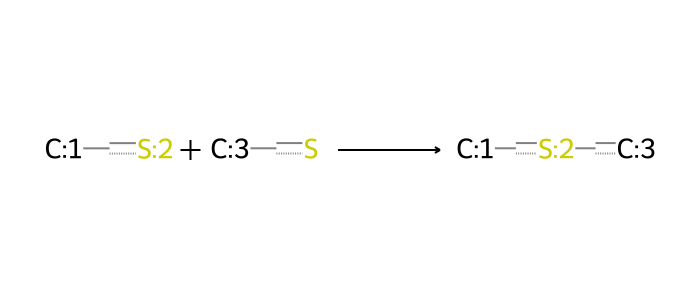
In synthesis mode, this illustrates a use for thiols as building-block fragments. It is therefore also useful to define other reactions that involve thiols. Thiol reactions with alkenes and alkynes fall into this category.
rxn_smarts_dict["thiol-yne"] = (
"[CX2H1:1]#[CX2:2]."
"[SX2H1:3]"
">>"
"[CX3H2:1]=[C:2][SX2:3]"
)
rxn_smarts_dict["thiol-ene"] = (
"[#6:1][CX3:2]=[CX3H2:3]."
"[SX2H1:4]"
">>"
"[#6:1][CX3H2:2][CX3H2:3][SX2:4]"
)
bio = io.BytesIO(ccd_tools.draw_reaction(rxn_smarts_dict["thiol-ene"]))
Image.open(bio)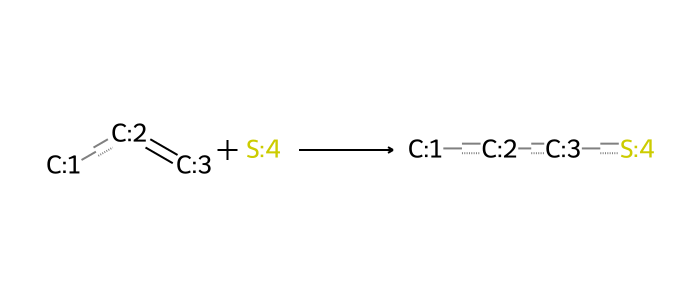
We could go on enumerating reactions here, including others with more than two reactants. But the iterative decomposition and synthesis routines provided in this post series do not handle reactions with than two reactants at present. In any case, such reactions can be rewritten in terms of multiple two-reactant reactions.
There are many examples of such reactions, like the one-pot Ugi 3-component reaction from the AGILE study (Xu et al. 2024):
ugi_str = "[#6:1][NX3:5]([H]).[#6:2][CX3H1:6](=O)[#6:3].[CX1-:7]#[N+:8][#6:4]>>[#6:1][N:5][C:6]([#6:2])([#6:3])[C+0:7](=[O])[N+0:8]([H])[#6:4]"
bio = io.BytesIO(ccd_tools.draw_reaction(ugi_str))
Image.open(bio)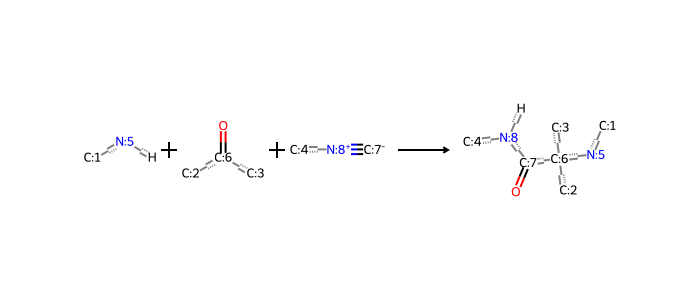
Many more examples of these and other reactions can be found in the literature (Hartenfeller et al. 2011), or taken from libraries of commercial interest, like this example.
We’ve compiled a fairly comprehensive set comprising the majority of known named ILs of commercial relevance in drug development. This is procured from purchasable LNP catalogs in another post.
import pandas as pd
all_structures = pd.read_csv(file_pfx + 'LNP_catalog/' + 'known_IL_combined.tsv', sep='\t', header=None, names=['SMILES', 'catalog', 'name'])
all_structures| SMILES | catalog | name | |
|---|---|---|---|
| 0 | C=O.C=O.CC.CCCCCCCCCCCC(O)CNCCN(CCN)CCN.CCCCCC... | ../../files/LNP_catalog/Medchem | Oligomer-lipid 7C1 |
| 1 | CC(C)=CCCC(C)CCSCC(C)C(=O)OCCOC(=O)CCN(CCCN(C)... | ../../files/LNP_catalog/Medchem | 4A3-Cit |
| 2 | CC(C)CCCC(C)CCCC(C)CC(=O)OCC(COC(=O)CCCN(C)C)(... | ../../files/LNP_catalog/Broadpharm | BP Lipid 517 |
| 3 | CC(C)CCCC(C)CCCC(C)CC(=O)OCC(COC(=O)CCN1CC(O)C... | ../../files/LNP_catalog/Broadpharm | BP Lipid 533 |
| 4 | CC(C)CCCC(C)CCCC(C)CC(=O)OCC(COC(=O)CCN1CCCC1)... | ../../files/LNP_catalog/Broadpharm | BP Lipid 521 |
| ... | ... | ... | ... |
| 552 | CCCCSSCCCCCCOC(=O)CCNCCCN(C)CCCN(CCC(=O)OCCCCC... | ../../files/LNP_catalog/Medchem | Lipid 9 |
| 553 | NCCN1CCN(CCN)CC1 | ../../files/LNP_catalog/Medchem | 2,2'-(Piperazine-1,4-diyl)diethanamine |
| 554 | [H]N(C(=O)C(CCCCCOC(=O)CCCCCCC/C=C\CCCCCCCC)N(... | ../../files/LNP_catalog/Broadpharm | 119-23 |
| 555 | [H]N(C(=O)CCCCC(CCSCCC(=O)OCCCCCC(C)C)SCCC(=O)... | ../../files/LNP_catalog/Broadpharm | CP-LC-1428 |
| 556 | [H]N(CCN(C)C)C(=O)C(CCSCCC(=O)OCCCCCCCCCCCCCCC... | ../../files/LNP_catalog/Broadpharm | CP-LC-1074 |
557 rows × 3 columns
This is a set of hundreds of structures comprising published ionizable lipids from various research programs. The predecessor post demonstrated decomposition of these molecules at their ester linkages, observing that this leads to relatively few distinct fragments that are each relatively light.
If we add more reactions, the fragments will be strictly less heavy, as some intermediate structures which would have been fragments under the previous reaction set are cleaved further. This is worthwhile if it leads to more shared fragments between molecules, showing the fragment-based redundancy in explored IL space. We use this realization to settle on a small set of reactions to decompose any set of chemical structures.
As in the previous post, we can demonstrate the new functionality by plotting how many fragments the known IL structures have, using different reaction decompositions.
rxn_smarts_dict{'esterification': '[#6:4][C:2](=[O:3])[OX2H1:1].[#6X4,c;!$(C([OX2H])[O,S,#7,#15]):6][OX2H1]>>[#6:6][O:1][C:2](=[O:3])[#6:4]',
'amide_coupling': '[#6:1][C:2](=[O:3])[OX2H1].[#6:6][NX3;H2;!$(NC=O):5]>>[#6:6][NX3R0;H1:5][CR0:2](=[O:3])[#6:1]',
'michael_addition_acrylate_1o': '[CX3H2:1]=[CH1:2][C:3](=[O:4])[O:5].[NX3;H2:6]>>[NX3;H1:6][CH2:1][CH2:2][C:3](=[O:4])[O:5]',
'michael_addition_acrylate_2o': '[CX3H2:1]=[CH1:2][C:3](=[O:4])[O:5].[NX3;H1:6]>>[NX3;H0:6][CH2:1][CH2:2][C:3](=[O:4])[O:5]',
'michael_addition_acrylamide_1o': '[CX3H2:1]=[CH1:2][C:3](=[O:4])[N:5].[NX3;H2:6]>>[NX3;H1:6][CH2:1][CH2:2][C:3](=[O:4])[N:5]',
'michael_addition_acrylamide_2o': '[CX3H2:1]=[CH1:2][C:3](=[O:4])[N:5].[NX3;H1:6]>>[NX3;H0:6][CH2:1][CH2:2][C:3](=[O:4])[N:5]',
'epoxide_ring_opening_1o': '[#6:1][CR1:4]1[CR1:3][OR1:2]1.[#6:6][NX3;H2;!$(NC=O):5]>>[#6:1][CR0:4]([OX2H1:2])[CR0:3][NX3;H1;!$(NC=O):5][#6:6]',
'epoxide_ring_opening_2o': '[#6:1][CR1:4]1[CR1:3][OR1:2]1.[#6:6][NX3;H1;!$(NC=O):5]>>[#6:1][CR0:4]([OX2H1:2])[CR0:3][NX3;H0;!$(NC=O):5][#6:6]',
'alcohols_to_ether': '[#6:1][OX2H1:2].[#6:3][OX2H1]>>[#6:1][OX2H0R0:2][#6:3]',
'thiol_to_disulfide': '[SX2H1:1].[SX2H1:2]>>[SX2H0:1][SX2H0:2]',
'thiols_to_thioether': '[#6:1][SX2H1:2].[#6:3][SX2H1]>>[#6:1][SX2H0:2][#6:3]',
'thiol-yne': '[CX2H1:1]#[CX2:2].[SX2H1:3]>>[CX3H2:1]=[C:2][SX2:3]',
'thiol-ene': '[#6:1][CX3:2]=[CX3H2:3].[SX2H1:4]>>[#6:1][CX3H2:2][CX3H2:3][SX2:4]'}The new reactions have to be given reaction codes, which are unique identifiers for the reactions that allow the decomposition to be expressed and reconstructed.
rxn_codes = {
"esterification": "R1",
"amide_coupling": "R2",
"michael_addition_acrylate_1o": "R3a",
"michael_addition_acrylate_2o": "R3b",
"michael_addition_acrylamide_1o": "R4a",
"michael_addition_acrylamide_2o": "R4b",
"epoxide_ring_opening_1o": "R5a",
"epoxide_ring_opening_2o": "R5b",
"alcohols_to_ether": "R6",
"thiol_to_disulfide": "R7",
"thiols_to_thioether": "R8",
"thiol-yne": "R9",
"thiol-ene": "R10"
}We can see the effect of varying the reaction set by illustrating using a lipid with a few heterogeneous linkages. Something like this IL from the catalog exhibits not only the common ester linkages, but also a few amide and thioether linkages.
IL_name = "CP-LC-1428"
test_smiles = all_structures["SMILES"][all_structures["name"] == IL_name].values[0]
Chem.MolFromSmiles(test_smiles)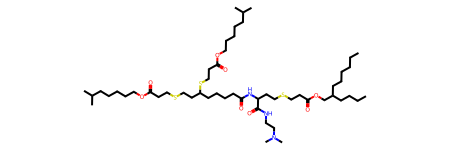
Cleaving the ester linkages cuts off some low-complexity alkyl tails which are ripe for further variation and screening. But with this decomposition, the core fragment is a large high-complexity moiety that goes far beyond the head; it is held together by a few amide and thioether linkages, which are not cleaved.
import ccd_tools
import mols2grid
decomp_results = ccd_tools.decompose_dataset(
[test_smiles],
rxn_smarts_dict,
rxn_codes,
reaction_types=["esterification"],
print_debug=False
)
smiles_display = decomp_results[2]
#names_display = ["Original"] + [rxn_codes[x] for x in decomp_results[1]]
mols2grid.display(
pd.DataFrame({
'SMILES': smiles_display,
#'numuq': ["In {} unique lipids".format(x) for x in sorted_frag_df['Number_of_unique_lipids']]
}),
#subset=['img', 'numuq'],
subimgsize=(600, 300)
)This is somewhat of an obstacle to easily varying the head group structure. To attain more control over the structure, we can use a more comprehensive reaction set that cleaves amide and thioether linkages as well.
This results in a set of much lighter fragments.
import ccd_tools
import mols2grid
decomp_results = ccd_tools.decompose_dataset(
[test_smiles],
rxn_smarts_dict,
rxn_codes,
reaction_types=["esterification", "amide_coupling", "thiol_to_disulfide", "thiols_to_thioether"],
print_debug=False
)
smiles_display = decomp_results[2]
#names_display = ["Original"] + [rxn_codes[x] for x in decomp_results[1]]
mols2grid.display(
pd.DataFrame({
'SMILES': smiles_display
})
)As a general matter, different reaction sets yield different sets of fragments. Expanding the reaction set always yields a higher number of fragments, and the fragments are less heavy than before. But they are not necessarily more or less diverse.
all_smiles = list(all_structures['SMILES'])
rxn_sets = {
"Ester cleavage only": ["esterification"],
"Ester and sulfide": ["esterification", "thiol_to_disulfide", "thiols_to_thioether"],
"Ester and amide cleavage": ["esterification", "amide_coupling"],
"Ester+amide+Michael": [
"esterification", "amide_coupling",
"michael_addition_acrylamide_1o", "michael_addition_acrylamide_2o",
"michael_addition_acrylate_1o", "michael_addition_acrylate_2o"
],
"Ester+amide+Michael+epoxide": [
"esterification", "amide_coupling",
"michael_addition_acrylamide_1o", "michael_addition_acrylamide_2o",
"michael_addition_acrylate_1o", "michael_addition_acrylate_2o",
"epoxide_ring_opening_1o", "epoxide_ring_opening_2o"
],
"Ester+amide+sulfide": ["esterification", "amide_coupling", "thiol_to_disulfide", "thiols_to_thioether"],
"All": list(rxn_smarts_dict.keys()),
}
decomp_info = {}
for name, rxn_set in rxn_sets.items():
decomp_info[name] = ccd_tools.decompose_dataset(
all_smiles, rxn_smarts_dict, rxn_codes,
reaction_types=rxn_set,
print_debug=False
)
print("{}: {} distinct fragments represent {} structures. ".format(
name, len(decomp_info[name][2]), len(all_smiles)))Ester cleavage only: 429 distinct fragments represent 557 structures.
Ester and sulfide: 433 distinct fragments represent 557 structures.
Ester and amide cleavage: 450 distinct fragments represent 557 structures.
Ester+amide+Michael: 441 distinct fragments represent 557 structures.
Ester+amide+Michael+epoxide: 426 distinct fragments represent 557 structures.
Ester+amide+sulfide: 454 distinct fragments represent 557 structures.
All: 413 distinct fragments represent 557 structures. As in the predecessor post, it’s interesting to plot the fragment counts and weights under different reaction sets.
import matplotlib.pyplot as plt
import numpy as np
import rdkit.Chem.Descriptors
plt.figure(figsize=(8, 4))
data_chemwts = [Chem.Descriptors.ExactMolWt(Chem.MolFromSmiles(x)) for x in all_smiles]
plt.hist(data_chemwts, bins=30, label=f'Lipids (n = {len(data_chemwts)})', alpha=0.5, density=True)
for s in rxn_sets:
input_lipid_desc, molecule_info_program, frags_program = ccd_tools.decompose_dataset(
all_smiles, rxn_smarts_dict, rxn_codes,
reaction_types=rxn_sets[s])
lipid_tags_df, frag_count_df, rxn_codes_df = ccd_tools.compute_fragment_counts(input_lipid_desc, frags_program, rxn_codes)
frag_count_df.set_index('SMILES', inplace=True)
newcols = list(frag_count_df.columns)
for i in range(3, frag_count_df.shape[1]):
sndx = np.where(np.array(all_smiles) == frag_count_df.columns[i])[0]
if len(sndx) > 0:
newcols[i] = all_structures['name'][sndx[0]]
frag_count_df.columns = newcols
newrows = list(frag_count_df.index)
for i in range(frag_count_df.shape[0]):
sndx = np.where(np.array(all_smiles) == frag_count_df.columns[i])[0]
if len(sndx) > 0:
newcols[i] = all_structures['name'][sndx[0]]
frag_count_df.index = newrows
data_fragwts = [Chem.Descriptors.ExactMolWt(Chem.MolFromSmiles(x)) for x in frags_program]
plt.hist(data_fragwts, bins=30, label=f'{s} (n = {len(data_fragwts)})', alpha=0.5, density=True)
plt.xlabel("Molecular weight")
plt.title(f"Distribution of molecular weights")
plt.legend()
plt.show()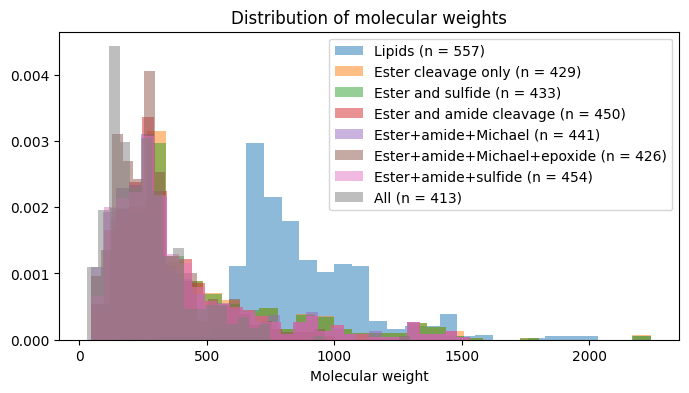
import matplotlib.pyplot as plt
input_lipid_desc, molecule_info_program, frags_program = decompose_dataset(all_smiles, rxn_smarts_dict, reaction_types=['esterification'])
lipid_tags_df, frag_count_df, rxn_codes_df = compute_fragment_counts(input_lipid_desc, frags_program, rxn_codes)
frag_count_df.set_index('SMILES', inplace=True)
newcols = list(frag_count_df.columns)
for i in range(3, frag_count_df.shape[1]):
sndx = np.where(np.array(all_smiles) == frag_count_df.columns[i])[0]
if len(sndx) > 0:
newcols[i] = all_structures['name'][sndx[0]]
frag_count_df.columns = newcols
newrows = list(frag_count_df.index)
for i in range(frag_count_df.shape[0]):
sndx = np.where(np.array(all_smiles) == frag_count_df.columns[i])[0]
if len(sndx) > 0:
newcols[i] = all_structures['name'][sndx[0]]
frag_count_df.index = newrows
import rdkit.Chem.Descriptors
data_chemwts = [Chem.Descriptors.ExactMolWt(Chem.MolFromSmiles(x)) for x in all_smiles]
data_fragwts = [Chem.Descriptors.ExactMolWt(Chem.MolFromSmiles(x)) for x in frags_program]
data1 = data_chemwts + data_fragwts
lbls1 = ['structures']*len(data_chemwts) + ['fragments']*len(data_fragwts)
def plot_hist_molwts(data1, data2, fam_name):
plt.figure(figsize=(8, 4))
plt.hist(data1, bins=30, label=f'Lipids (n = {len(data1)})', alpha=0.5, density=True)
plt.hist(data2, bins=30, label=f'Fragments (n = {len(data2)})', alpha=0.5, density=True)
plt.xlabel("Molecular weight")
plt.title(f"{fam_name}: Distribution of molecular weights")
plt.legend()
plt.show()
plot_hist_molwts(data_chemwts, data_fragwts, 'All lipids')import matplotlib.pyplot as plt
plt.hist([len(ccd_tools.decompose_into_smiles(x, e1_molecule_info)) for x in all_smiles])
plt.xlabel("Number of fragments per molecule")
plt.ylabel("Number of molecules")
plt.title("Fragmentation of molecules by ester cleavage")
plt.show()We have seen that the reaction set can have a significant effect on the fragments that are obtained. This is a key consideration when considering fragment libraries for combinatorial chemistry and ultralarge virtual screening spaces.
For the particular example of ionizable lipids in LNPs, we can also see that there are essentially a few hundred possible fragments covered by the literature. This compares to thousands of fragments for generic organic molecule classes like acids and amines, which are available from similar purchasable catalogs. This orders-of-magnitude difference between the LNP literature and purchasable catalogs indicates the specificity of the LNP molecule class, and points to the possibilities of further expanding virtual spaces for exploration.
For future such reference, it’s useful to collect all the code from this post in a .py file.
all_code = r'''rxn_smarts_dict = {}
rxn_smarts_dict["esterification"] = (
"[#6:4][C:2](=[O:3])[OX2H1:1]."
"[#6X4,c;!$(C([OX2H])[O,S,#7,#15]):6][OX2H1]"
">>"
"[#6:6][O:1][C:2](=[O:3])[#6:4]"
)
rxn_smarts_dict["amide_coupling"] = (
"[#6:1][C:2](=[O:3])[OX2H1]."
"[#6:6][NX3;H2;!$(NC=O):5]"
">>"
"[#6:6][NX3R0;H1:5][CR0:2](=[O:3])[#6:1]"
)
rxn_smarts_dict["michael_addition_acrylate_1o"] = (
"[CX3H2:1]=[CH1:2][C:3](=[O:4])[O:5]."
"[NX3;H2:6]"
">>"
"[NX3;H1:6][CH2:1][CH2:2][C:3](=[O:4])[O:5]"
)
rxn_smarts_dict["michael_addition_acrylate_2o"] = (
"[CX3H2:1]=[CH1:2][C:3](=[O:4])[O:5]."
"[NX3;H1:6]"
">>"
"[NX3;H0:6][CH2:1][CH2:2][C:3](=[O:4])[O:5]"
)
rxn_smarts_dict["michael_addition_acrylamide_1o"] = (
"[CX3H2:1]=[CH1:2][C:3](=[O:4])[N:5]."
"[NX3;H2:6]"
">>"
"[NX3;H1:6][CH2:1][CH2:2][C:3](=[O:4])[N:5]"
)
rxn_smarts_dict["michael_addition_acrylamide_2o"] = (
"[CX3H2:1]=[CH1:2][C:3](=[O:4])[N:5]."
"[NX3;H1:6]"
">>"
"[NX3;H0:6][CH2:1][CH2:2][C:3](=[O:4])[N:5]"
)
rxn_smarts_dict["epoxide_ring_opening_1o"] = (
"[#6:1][CR1:4]1[CR1:3][OR1:2]1."
"[#6:6][NX3;H2;!$(NC=O):5]"
">>"
"[#6:1][CR0:4]([OX2H1:2])[CR0:3][NX3;H1;!$(NC=O):5][#6:6]"
)
rxn_smarts_dict["epoxide_ring_opening_2o"] = (
"[#6:1][CR1:4]1[CR1:3][OR1:2]1."
"[#6:6][NX3;H1;!$(NC=O):5]"
">>"
"[#6:1][CR0:4]([OX2H1:2])[CR0:3][NX3;H0;!$(NC=O):5][#6:6]"
)
rxn_smarts_dict["alcohols_to_ether"] = (
"[#6:1][OX2H1:2]."
"[#6:3][OX2H1]"
">>"
"[#6:1][OX2H0R0:2][#6:3]"
)
rxn_smarts_dict["thiol_to_disulfide"] = (
"[SX2H1:1]."
"[SX2H1:2]"
">>"
"[SX2H0:1][SX2H0:2]"
)
rxn_smarts_dict["thiols_to_thioether"] = (
"[#6:1][SX2H1:2]."
"[#6:3][SX2H1]"
">>"
"[#6:1][SX2H0:2][#6:3]"
)
rxn_smarts_dict["thiol-yne"] = (
"[CX2H1:1]#[CX2:2]."
"[SX2H1:3]"
">>"
"[CX3H2:1]=[C:2][SX2:3]"
)
rxn_smarts_dict["thiol-ene"] = (
"[#6:1][CX3:2]=[CX3H2:3]."
"[SX2H1:4]"
">>"
"[#6:1][CX3H2:2][CX3H2:3][SX2:4]"
)
rxn_codes = {
"esterification": "R1",
"amide_coupling": "R2",
"michael_addition_acrylate_1o": "R3a",
"michael_addition_acrylate_2o": "R3b",
"michael_addition_acrylamide_1o": "R4a",
"michael_addition_acrylamide_2o": "R4b",
"epoxide_ring_opening_1o": "R5a",
"epoxide_ring_opening_2o": "R5b",
"alcohols_to_ether": "R6",
"thiol_to_disulfide": "R7",
"thiols_to_thioether": "R8",
"thiol-yne": "R9",
"thiol-ene": "R10"
}
'''file_pfx = "../../files/chem/"
with open(file_pfx + "db_reactions.py", "w", encoding="utf-8") as f:
f.write(all_code)@online{balsubramani,
author = {Balsubramani, Akshay},
title = {Decomposition and Synthesis with a Flexible Reaction Set},
langid = {en}
}