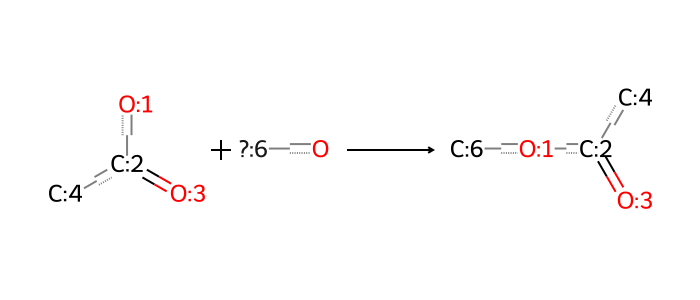Some of the most exciting algorithmic advances in applied drug discovery involve manipulating chemical spaces that are truly vast. When the space cannot be sampled and cleverly stored in memory, the modular structure relating chemicals in the space to each other is key to concisely manipulating and learning on it.
The ubiquitous cheminformatics package RDKit has some wonderful functionality for searching over synthon spaces that are represented in this manner. The functionality runs blazing fast, even on inconceivably large virtual spaces, because it does not scale in the combinatorics of putting the synthons together, only in the number of distinct synthons themselves.
Many synthon formats have blossomed in the last few years. Most of the time, they are equivalent to each other, finding different ways of encoding fragments and how to put them together.
Here we collect some useful code for dealing with these formats and manipulating them. We show how some of this functionality will be useful, in a way that’s complementary to existing canonical demonstrations of it, working in the RDKit ecosystem as usual.
Synthons in standard form
Representing spaces implicitly with synthons
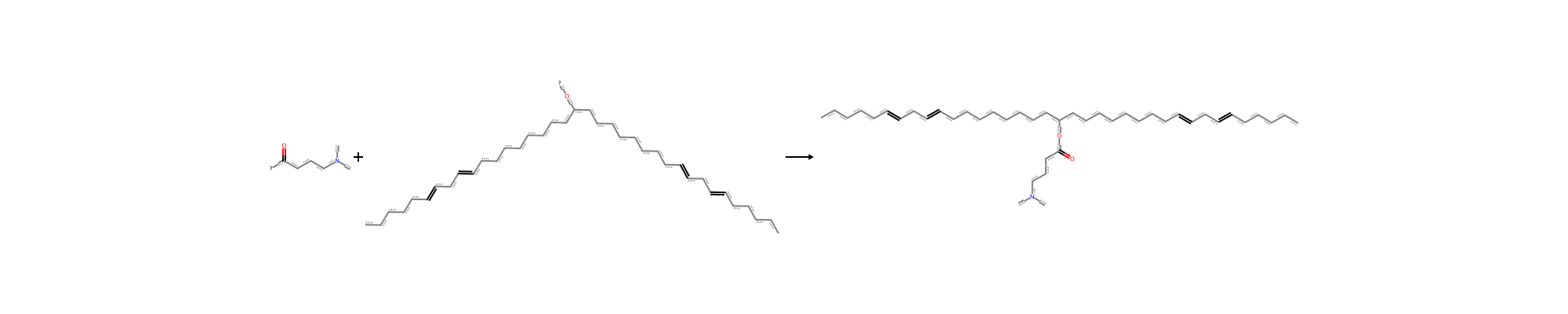
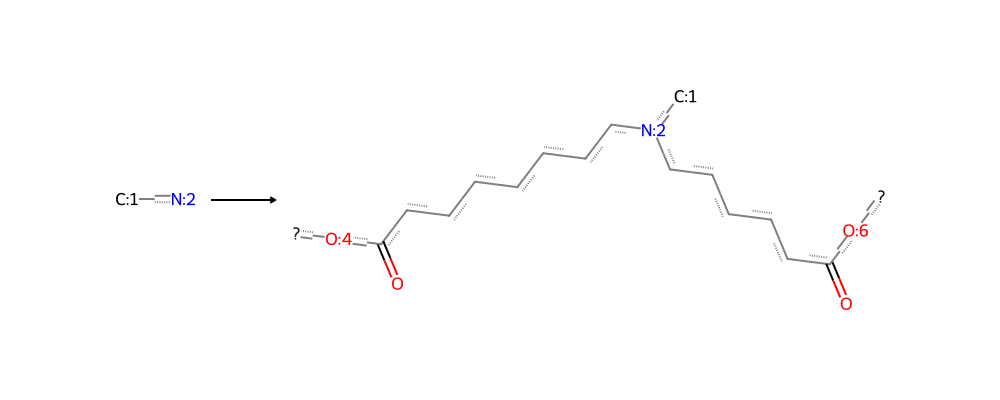
A synthon is a convenient unit of retrosynthetic analysis in chemistry. For us it has a precise computational meaning – a synthon is a fragment with reactive centers indicated by dummy atoms, which are removed when an in silico bond formation (“reaction”) occurs. Here is a simple example, a bond formation between two reactants, namely the ester linkage of the ionizable lipid MC3.
On one hand, this representation is unambiguous. Given two synthons, I know exactly how to put them together without any further information.
On the other hand, this way of representing synthons is not synthetically plausible as it does not seek to mimic any physical reaction mechanism. All carbons and heteroatoms are conserved, and only bonds between existing atoms are formed.
Putting these synthons together in a file is a beautifully concise way of representing a space. A synthon file is a file with one synthon per line, in a very specific format which we review below because of the scarcity of other documentation.
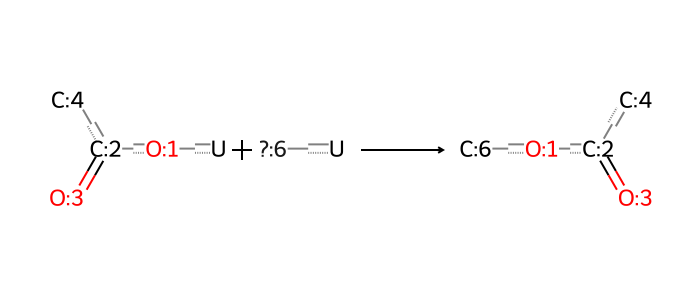
Each synthon has at least one attachment point to other synthons, which allows it to participate in in silico reactions.
Column name
Description
Notes
SMILES
Synthon SMILES with attachment atoms marked as [1*], [2*], … (or the transuranic labels [U], [Np], [Pu], [Am] for up to 4).
Required
synton_id
String identifier, e.g. 3-1.
Required
synton#
Integer role (0, 1, 2, …) within its reaction.
Required
reaction_id
Name/ID grouping synthons into one reaction.
Required
release
Free-text “release” tag.
Optional
A collection of synthons combinatorially describes a space according to the following parsing rule.
For each reaction_id, take one synthon for each synton# present, giving a set of multiple synthons.
Replace any pairs of matching attachment labels (e.g. [1*], [2*], …) by a direct bond between the linked atoms.
Summary: RDKit synthon file format
Header line must literally read SMILES synton_id synton# reaction_id
or (with optional column) SMILES synton_id synton# reaction_id release
(synton spelling is deliberate).
Separator:
Tab by default, any ASCII whitespace accepted.
A comma-separated variant is also allowed (SMILES,synton_id,synton_role,reaction_id,…).
Validation: loader checks header and correct field count per row; else throws std::runtime_error.
Interrupts: if the process receives SIGINT, the reader exits early with cancelled = true.
Synthons from reaction chemistry
Conventionally, virtual spaces are generated by applying in silico SMARTS reactions to realistic fragments in ways that mimic known chemistry. As I’ve written, such reactions and fragments can be generated for known small-molecule design spaces, such as ionizable lipid design for LNP engineering – for instance, carboxylic acid fragments and alcohols can combine to make ester linkages. This approach leads to virtual spaces which tend to have more developable and otherwise synthesizable molecules, by systematically generating variations on existing reaction schemas.
In such situations, we may want to take advantage of fast synthon-based functionality provided by RDKit. Doing this is a non-trivial process which uses knowledge of the underlying chemistry. Here we will work through how it can be done for an example of interest – LNP engineering.
Examining a reaction
Overall, this example illustrates how to convert any reaction schema – comprised of lists of fragments and reaction SMARTS combining them – into a synthon file. Each reaction is used to build synthons under its reaction_id. The number of reactants is reflected in the maximum synton# for that reaction_id. For each such reactant, possible synthons are appended to the synthon file by referring to the fragment files.
The starting point is the reactions used to comprise the synthon space. We’ll start with a single reaction for simplicity. Let’s take the esterification reaction, which we discuss in another post on LNP engineering, as a prime example. This reaction is, as written, a coupling between an acid and an alcohol that involves dehydration.
But the dehydration involves removal of a heteroatom, and therefore won’t fit in the RDKit synthon schema. Instead, the heteroatom must be taken out of one of the reactants. In this case, the natural chemical choice is the alcohol, leading to a reaction that does fit into the RDKit synthon schema. (Recall that the attachment points are labeled with transuranics [U], [Np], etc as specified above.)
This dictates how we assemble the synthons. The reaction is asymmetric, and the reactants play asymmetric roles. So we should always think carefully about the chemical intuition behind associating a certain set of synthons with a reactant.
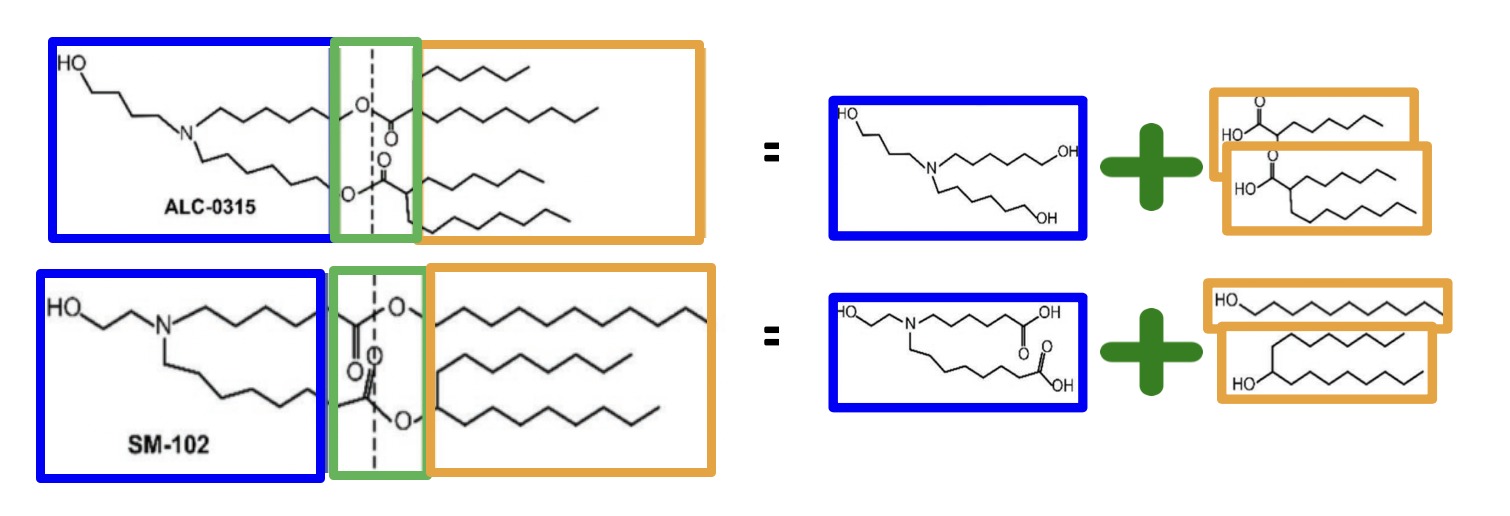
By this reasoning, we realize that the above esterification reaction is not enough to fully specify the synthons. Consider, for instance, two different occurrences of esterification, in the ionizable lipids ALC-0315 and SM-102:
Figure 1: Adapted from Jorgensen et al. 2023
If we’re considering ALC-0315, the acid synthons correspond to lipophilic tails, and the alcohol synthons to ionizable heads. On the other hand, if we’re considering SM-102, it is the alcohol synthons that are the tails, while the acid synthons correspond to ionizable heads. This starkly demonstrates how the synthon definition depends on the context of the synthesis being considered, not just on the synthesis reaction itself.
Outline
So let’s specify our demonstration scenario in more detail.
We’ll demonstrate making variations on SM-102, the same situation as outlined in another blog post on scoring ultralarge spaces. The scenario involves modifying the amine ionizable head moiety and the lipophilic alcohol tail moieties, while maintaining the two-tailed structure and spacer carbons between them.
We’ll use synthons derived from the head and tail fragments used there, with the reactive sites at the ester linkages only.
Assembling synthons for a reaction
Now we focus on the key conversion from fragments into synthons, ranging over the reactants in the above esterification reaction and assembling possible synthons for each. This is not trivial – in addition to removing heteroatoms, we must also mark reaction sites. This requires us to look up new information which is left technically ambiguous in the standard way of representing reactions.1
This task is an opportunity for useful insight into the chemistry of these virtual spaces. We range over the fragments and detect appropriate chemical moieties (acid groups, alcohol groups, etc.) to determine which fragments can participate in which roles, as synthons in the reaction.
Acid synthons
According to the esterification reaction outlined above, the first reactant should be a carboxylic acid, with two acid moieties that will form ester linkages with tails. As explained further in the other post on ultralarge scoring, we can create variants of SM-102 by varying this head fragment systematically according to a comprehensive list of thousands of primary amines known in the literature. We substitute each of the hydrogens on the primary amine with an alkyl spacer chain leading to an acid reactive moiety, which will join with a tail synthon. This substitution reaction results in a set of synthons, corresponding to the first (acid) reactant in the esterification.
import pandas as pdhead_pfx ="../../files/LNP_catalog/"df = pd.read_csv(head_pfx +'frags_amines_TS.csv.gz', compression='gzip')df
SMILES
Name
Amine_type
0
C1CNC2NCCNC2N1
Whitehead_0
secondary_amine | secondary_amine | secondary_...
1
C1CNCCCNCCCNC1
Whitehead_1
secondary_amine | secondary_amine | secondary_...
2
C1CNCCCNCCNCCCNC1
Whitehead_2
secondary_amine | secondary_amine | secondary_...
3
C1CNCCN1
Whitehead_3
secondary_amine | secondary_amine
4
C1CNCCNC1
Whitehead_4|CSSS00000210015
secondary_amine | secondary_amine
...
...
...
...
39531
N#CCn1nnc(N)n1
Klarich_34290744
primary_amine | aromatic_pyridinic | aromatic_...
39532
Nc1nnc2nccnn12
Klarich_2289036
primary_aniline | aromatic_pyridinic | aromati...
39533
CNC(=O)n1nnnc1N
Klarich_5510474
secondary_amine | aromatic_pyridinic | aromati...
39534
Nc1ncc2n[nH]nc2n1
Klarich_18562019
primary_aniline | aromatic_pyridinic | aromati...
39535
N=C(N)Nc1nnn[nH]1
Klarich_4404328
primary_amine | primary_amine | secondary_anil...
39536 rows × 3 columns
Alcohol synthons
Synthons for the alcohol reactants are easy to determine using the reasoning we’ve outlined. Each new bond simply attaches to a hydroxyl carbon, i.e. the \(\alpha\) carbon of the alcohol.