AI in the small-molecule discovery funnel
Summary
TL;DR: This article implements value-adding steps in small-molecule drug design, demonstrated on the running example of lipid nanoparticle (LNP) drug discovery. This can be considered a summary of a collection of blog posts, which contain implementations and explanations of useful cheminformatic functions to conceive, construct, and work with ultralarge chemical spaces.
Sourcing existing chemical structures
Working with more structures than we can count
Above and beyond a single program
There are other posts that also may be useful here, mostly those on chemistry-oriented data visualization, like:
AI in modern drug discovery pipelines
Modern drug discovery increasingly relies on machine learning (ML) and AI techniques to search over vast chemical spaces and expedite the design of novel therapeutics. A contemporary drug discovery pipeline often interweaves many key AI-driven components, among them prediction models, molecular encoders (embeddings), and molecular decoders (generators).
Components of a pipeline
In broad terms, one can imagine an iterative loop: candidate molecules are proposed (via a generative model or library search, i.e. a decoder), evaluated for desired properties (via predictive models and wet-lab experiments), and refined using learned representations of molecular structure (via encoders that produce latent embeddings). These steps enable both the exploration of chemical space (through generation of new candidates) and the exploitation of learned structure–activity relationships (through property prediction on those candidates). In such a pipeline,
- A prediction model might be a quantitative structure–activity relationship (QSAR) model or a neural network that estimates, for example, a drug’s potency, ADME profile, or another measure of efficacy or toxicity of a formulation.
- An encoder is typically a model (like a graph neural network or a language model for SMILES) that converts a molecule into a latent vector or embedding which captures salient chemical features.
- A decoder is the inverse: a method to go from an abstract representation back to a concrete molecular structure. Decoders can be explicit generative models (such as variational autoencoders or transformers that output molecular graphs/SMILES) or even simple lookup procedures into a predefined virtual library.
If we have a way to traverse the model’s latent representation and generate molecules (via a decoder), we can ask the model to suggest new lipids that work best according to the prediction model.
This is the essence of AI-guided design: a learned embedding space of molecules can be searched and optimized over in silico, and the results are decoded to actual structures to try in the lab.
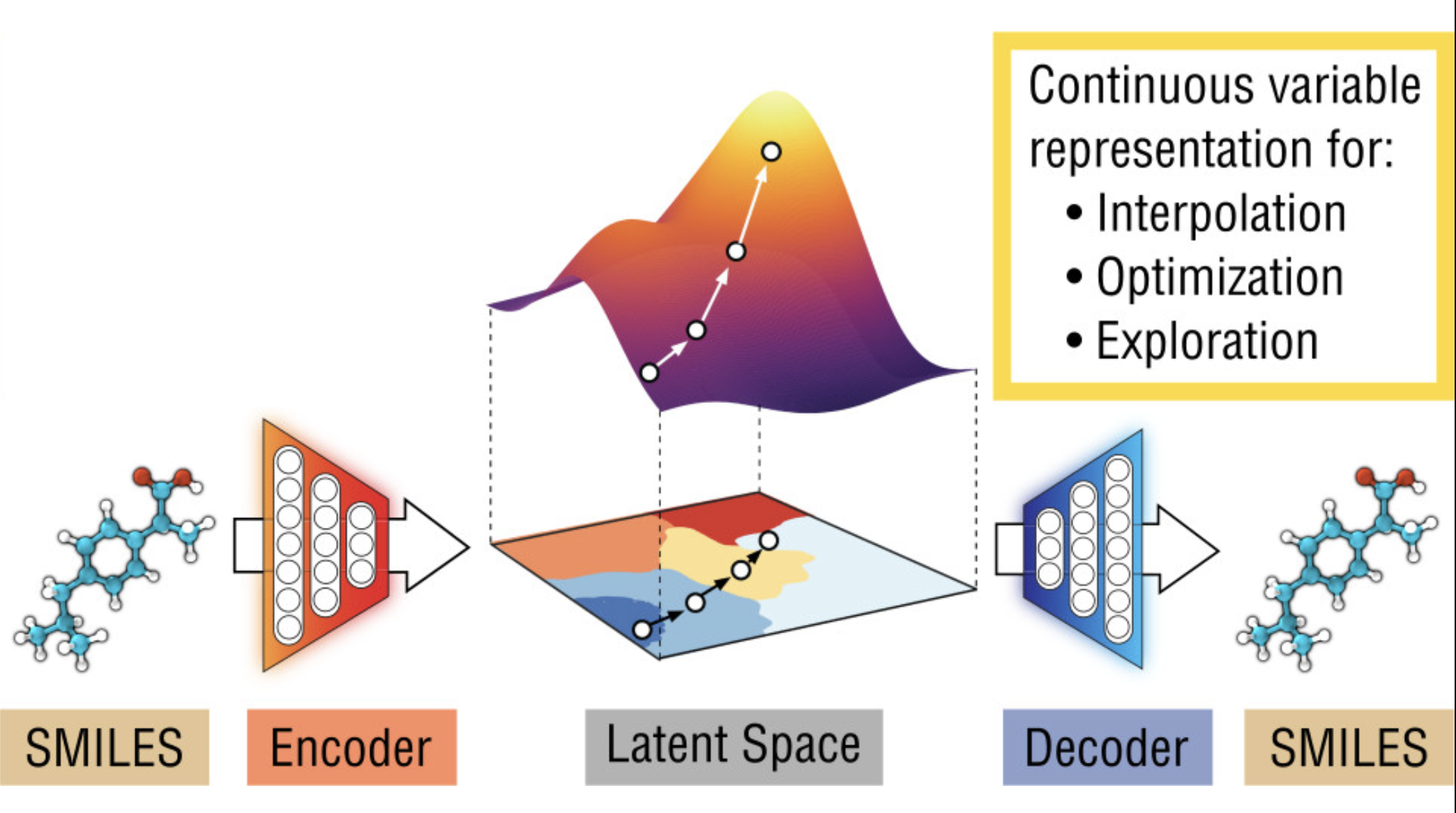
This overall approach has been at the foundation of the current wave of advances using AI to generate drug candidates (Gómez-Bombarelli et al. 2018).
Decodability and virtual spaces
Crucially, the pipeline’s success hinges on linking the learned representation space to actual synthesizable molecules — every point in the model’s latent space should correspond to a real compound that can be made or purchased. In other words, for any powerful molecular encoder we develop, we need a reliable molecular decoder to realize its insights in practice. Ensuring this alignment (sometimes termed validity or decodability of the latent space) is essential; without it, an AI model might predict an “ideal” compound that is chemically impossible or inaccessible. Recent advances explicitly address this by constraining generation to a known virtual space of molecules, or building blocks / reaction rules, so that any proposed molecule is by construction synthesizable. This is a particularly universal and transparently interpretable way to ensure that the model’s outputs are chemically valid – under names such as “maximum-likelihood decoding,” this strategy predominates, including in state-of-the-art design pipelines ((Gómez-Bombarelli et al. 2018), (Ross et al. 2022)). In traditional small-molecule drug discovery, one often starts with a large virtual library (millions to billions of molecules from databases like ZINC) or uses a generative model to propose new structures. This marriage of generative models with synthetic feasibility is transforming how we search chemical space, but is tricky to design around and implement correctly. In this series of posts, we go into such issues in detail and show exactly how to put together a robust in silico pipeline to apply predictive models on an unprecedented scale, using domain-specific chemistry knowledge to inform computational tools.
Engineering ionizable lipids for nanoparticle drug delivery
A prime example is the design of ionizable lipids for LNPs. Ionizable lipids (ILs) are a critical component of LNP formulations, which gained fame as the delivery vehicles for mRNA vaccines. Unlike conventional drug molecules that directly act on a biological target, ionizable lipids serve as enablers of delivery: they form nanoparticles that encapsulate nucleic acids (mRNA, siRNA, etc.), ferry them into target cells, and release them intracellularly. These lipids have a peculiar but essential trait: they are nearly neutral at physiological pH but cationic (positively charged) in mildly acidic environments such as the endosome, disrupting the endosomal membrane and allowing LNPs to escape the endosome while minimizing toxicity in the bloodstream or tissues at neutral pH. By condensing the negatively charged nucleic acids and then triggering endosomal release, ionizable lipids play a vital role in whether an LNP successfully delivers its cargo.
Designing ionizable lipids is a complex balancing act, and different therapeutic goals demand different lipid molecular designs. Some therapeutic applications, like gene editing in specific organs, might demand lipids that preferentially target certain cell types, have certain surface characteristics, or have a particular immunostimulatory profile. Achieving these disparate goals means tuning structural properties like the hydrophobic tail lengths (affecting lipid packing and clearance), the linker biodegradability (affecting persistence and toxicity), and the head group structure (affecting (pK_a) and ionization behavior). This modular view underpins many successful LNP lipid discovery efforts, including for the lipids used in the successful COVID-19 vaccines.
By swapping out these components, researchers have been creating new lipids with tunable properties. The diversity of possible head/linker/tail combinations is enormous, which presents both opportunities and challenges. There are opportunities to in principle tailor-make a lipid for a given therapeutic context (mRNA vaccines, liver-targeted siRNA, gene therapy to specific tissues, etc.), but challenges in finding suitable structures amid a combinatorial explosion of possibilities in the virtual space.
An AI-driven pipeline for ionizable lipid design can greatly accelerate this search by intelligently navigating a vast virtual space of candidates, focusing on those likely to meet the desired criteria for a given goal (e.g., high transfection efficiency in lung epithelial cells with low toxicity).
In the following sections, we outline a state-of-the-art pipeline for ionizable lipid engineering. This pipeline illustrates how modern AI and informatics tools can synergize to tackle the ionizable lipid design problem, from data gathering all the way to virtual screening of huge libraries.
Encoders, decoders, and virtual libraries
One of the first steps in any AI-driven molecular design workflow is to define the search space of candidate molecules. Often, when using an off-the-shelf end-to-end system, this is defined by the structures that the encoder has seen in training. However, many such off-the-shelf models (e.g. (De Cao and Kipf 2018), (Gómez-Bombarelli et al. 2018), (Irwin et al. 2022), (Haddad et al. 2025)) are trained on general-purpose chemical datasets, which do not typically include the specific classes of molecules we care about designing.
We can demonstrate this in detail when designing ionizable lipids for LNPs.
For ILs, especially novel ones, there is no single comprehensive database containing all plausible lipids. Therefore, an effective strategy is to construct a virtual chemical space of ionizable lipids by combining knowledge from literature and chemistry.
Complementary approaches are essential to do this well:
Aggregate known ionizable lipid structures (from papers, patents, catalogs), ensuring we learn from what has already been made and tested. This is covered in the post “Sourcing structures for in silico LNP discovery.”
Enumerate new structures via combinatorial chemistry rules. This allows exploration beyond known lipids by in silico synthesis of novel variants. As such, it’s vital to expanding the scope of any AI in these design pipelines.
- A basic toolkit is in the post “In silico fragment synthesis with combinatorial chemistry”.
- The flip side of this is decomposition: how the fragment composition of a structure can be determined, developed in the post “Combinatorial chemistry decompositions.”
Both approaches are greatly facilitated by reliable encoders/decoders. An AI model can learn an embedding from known lipids (encoder), but we also need the capability to decode a set of coordinates in that learned space into an actual chemical structure. Having an explicit library of candidates or a rule-based generator acts as that decoder, so that model suggestions correspond to synthesizable molecules. However, if the decoder is unreliable, and produces chemically implausible or difficult-to-synthesize structures, then the whole exercise fails. This is why many recent efforts, including specialized generative models for ionizable lipids, constrain generation to known structures, reaction schemes, or building blocks. By doing so, the idea is to make any point in latent space correspond to a combination of real fragments and reactions that yield a valid IL, grounding the virtual chemical space in reality.
The problem: too many structures to count
The challenge of defining a virtual chemical space is compounded by the staggering number of possible ionizable lipids. The combinatorial explosion of fragments makes it impossible to exhaustively enumerate all possible structures – even developable subsets routinely contain billions of candidates. In this environment, design teams hope to have a good starting point to winnow down the search space. In practice, though, trimming this to a manageable size involves unjustified assumptions and unsavory tradeoffs, without guaranteeing that the most promising candidates are included. This is where the power of AI comes in: it can help us navigate this vast space and identify promising candidates without needing to enumerate every possible structure, using understanding of the underlying chemistry to strike a careful balance between exploration and exploitation.
We show how to score the chemical space without enumerating it. This involves several complementary perspectives.
- Intelligently sampling the space: We do not need to consider every molecule to iteratively refine the model, using active learning techniques, training surrogate models, and so on.
- Using fragment-based scoring: The fragment-based structure can also be harnessed – as hinted earlier, if a property (like a contribution to a biological activity) can be decomposed into fragment contributions, we could score fragments instead of full molecules, then rapidly compute any combination’s score by aggregation. Such fragment-based machine learning models (such as message-passing neural networks that naturally consider parts) can generalize to new combinations without explicitly scoring each combination from scratch. This massively reduces the effective complexity of the space.
We show how to perform analysis over the chemical space without enumerating it, for common tasks like searching for substructures and nearest neighbors.
- Similarity and substructure search: An encoder that produces embeddings for molecules allows us to organize the space in a latent vector geometry. Similar molecules cluster together. If we find a region in embedding space that corresponds to good activity (say a cluster of known potent lipids), we can focus the search around that region, using distance metrics to find analogs. This approach, sometimes called nearest-neighbor search or clustering in chemical space, can narrow down candidates.
- Analog generation: The encoder-decoder combination can be used to search for analogues of an out-of-library molecule (perhaps a very potent or non-toxic lipid reported in a paper): encode it into the space, then search the latent space for the closest vectors that correspond to in-library (synthesizable) molecules.
All these implementations amplify the power of the base property prediction model, to predict over inconceivably large chemical spaces:

The future: tools for pattern recognizers
All this means that the role of AI in drug discovery is growing quickly. Already, drug development decisions are complex for unavoidable scientific and business reasons, and AI is being used to bring structure and data to bear on these decisions. The data being gathered are increasingly granular beyond unaided human comprehension, including genomic data representing highly integrative and multilayered translational biology, structured assays to measure downstream drug responses, the combinatorics of large chemical design spaces, and more.
This is complex, hard-won intellectual property that drug designers and decision-makers wish to use in a flexible way – a job well-suited to AI. Ideally, a human designer could set a provisional goal (e.g. “find me lipids suitable for pulmonary delivery [which might mean a certain range of (pK_a), high biodegradability, and strong endosomal release capacity]”), and the AI machinery would sift through the enormous virtual space to propose a short-list of candidates.
This challenge can be tackled with the tools and resources we write about here. These tools can power human and AI scientists and agents in a large variety of drug discovery scenarios, like in IL discovery. One example that we implement is trawling Google Scholar to find a comprehensive set of sources to retrieve further information from. This can be fed into AI agents.
We are overviewing a microcosm of the larger trend: use AI to supercharge the ability of natural scientists to find needles in haystacks – or perhaps to make new and larger haystacks and then find shinier needles within.
References
Reuse
Citation
@online{balsubramani,
author = {Balsubramani, Akshay},
title = {AI in the Small-Molecule Discovery Funnel},
langid = {en}
}